Acyanotic Lesions
Acyanotic diseases associated with normal pulmonary flow include those with systemic outflow obstruction such as coarctation of the aorta and aortic stenosis, mild and moderate pulmonary outflow stenosis, myocardial diseases, and arrhythmias. Acyanotic lesions usually associated with increased pulmonary blood flow include ventricular septal defect, atrial septal defect, endocardial cushion defects, patent ductus arteriosus, aortopulmonary window, and arteriovenous malformations (see Tables 33-14 and 33-15).
Acyanotic Anomalies with Systemic Outflow Obstruction
Coarctation and Interruption of the Aorta
For clinical, prognostic, and probably etiologic reasons, coarctation of the aorta is best considered in two separate categories: simple and complex. Simple coarctation is usually a discrete constriction of the aortic isthmus area, occasionally associated with a patent ductus arteriosus inserting just at or below it. Complex coarctation involves tubular hypoplasia of the aortic arch, with or without discrete aortic narrowing and one or more of the following lesions: patent ductus arteriosus, ventricular septal defect, endocardial cushion defect, aortic stenosis, sub-aortic stenosis, mitral stenosis or regurgitation, hypoplasia of the left ventricle and ascending aorta, other cyanotic anomalies, and endocardial fibroelastosis. In its most severe form the aortic arch may be atretic and completely interrupted as in DiGeorge syndrome. In complex coarctation and interruption of the aorta, the amalgam of left-sided involvement may be secondary to reduced intrauterine flow through the left heart, with consequent underdevelopment and hypoplasia extending from the left atrium to the aortic isthmus. In simple and complex coarctation, there are great variations possible in the extent and location of the coarctation.
Coarctation occurs in 6% to 7% of newborns with heart disease (see Tables 33-1 and 33-2). It is one of the common causes of congestive failure in the neonate (see Tables 33-7). Among symptomatic infants, 82% have complex coarctation, and 18% have simple coarctation. It is more common in male and premature infants. Girls sometimes have Turner
syndrome. Severe extra-cardiac anomalies, usually renal or gastrointestinal, occur in 6% to 9% of these patients (see Table 33-4).
syndrome. Severe extra-cardiac anomalies, usually renal or gastrointestinal, occur in 6% to 9% of these patients (see Table 33-4).
TABLE 33-14 FINDINGS IN ACYANOTIC 0- TO 2-WEEK-OLD NEONATES WITH CONGESTIVE HEART FAILUREa | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Pathophysiology
Simple Coarctation. The isthmus is normally smaller than the ascending or descending aorta in newborn infants because only 10% of the combined ventricular output during fetal life passes through the isthmus into the descending aorta, whereas approximately 60% passes through the ductus arteriosus to the descending aorta. After birth, the isthmus gradually grows, but in simple coarctation, there is a constricting band just above the point of connection to the ductus arteriosus. Aortic coarctation may acutely further constrict in the neonatal period, as constriction of the adjacent ductal tissue occurs. During infancy, there may be relative progression of the coarctation from inadequate,
disproportionately small aortic isthmus growth, and perhaps aortic wall hypertrophy and endothelial thickening at the coarctation site. Collateral circulation may be present at birth. In simple coarctation, the increased resistance to flow results in a pressure overload on the left ventricle. If the coarctation is not severe and there is a patent ductus arteriosus, with fall in pulmonary vascular resistance after birth, there is a reversal of flow through the ductus arteriosus from the aorta to the pulmonary artery, and a considerable left-to-right shunt may develop. If the increased pressure and volume load exceed the ability of the heart to compensate by hypertrophy or dilation, congestive failure with diminution of systemic output ensues. Left ventricular end-diastolic pressure is elevated, resulting in increased pulmonary venous pressure and development of pulmonary edema. The increased pulmonary venous pressure also produces pulmonary artery hypertension and right heart failure.
disproportionately small aortic isthmus growth, and perhaps aortic wall hypertrophy and endothelial thickening at the coarctation site. Collateral circulation may be present at birth. In simple coarctation, the increased resistance to flow results in a pressure overload on the left ventricle. If the coarctation is not severe and there is a patent ductus arteriosus, with fall in pulmonary vascular resistance after birth, there is a reversal of flow through the ductus arteriosus from the aorta to the pulmonary artery, and a considerable left-to-right shunt may develop. If the increased pressure and volume load exceed the ability of the heart to compensate by hypertrophy or dilation, congestive failure with diminution of systemic output ensues. Left ventricular end-diastolic pressure is elevated, resulting in increased pulmonary venous pressure and development of pulmonary edema. The increased pulmonary venous pressure also produces pulmonary artery hypertension and right heart failure.
TABLE 33-15 FINDINGS IN ACYANOTIC 2- TO 8-WEEK-OLD NEONATES WITH CONGESTIVE HEART FAILUREa | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Complex Coarctation and Aortic Interruption. Complex coarctation and aortic interruption are characterized by pulmonary artery hypertension with a ductus arteriosus supplying the descending aorta, usually a large intracardiac left-to-right shunt, and increased pulmonary flow. The right-sided structures are dilated and hypertrophied. There is a pressure and volume overload on both ventricles and congestive heart failure. In those with a large ventricular septal defect and patent ductus arteriosus, the systolic pressures in the pulmonary artery, descending aorta, ascending aorta, and right ventricle are identical. Peripheral pulse pressure is normal, and the pulses are equal throughout. With ductal constriction, the femoral arterial pulsations diminish. If the aortic arch obstruction is severe or complete, perfusion to the lower one-half of the body, previously supplied by the open ductus, is reduced. Manifestations of shock, renal and mesenteric hypoperfusion, and metabolic acidosis develop. Ductus closure causes death.
Clinical Findings
Simple Coarctation. Infants with isolated discrete coarctation may be asymptomatic, although some develop congestive heart failure, often after the age of 1 month. The femoral and pedal pulses are absent or diminished compared with brachial or carotid pulses. Radial and brachial pulses may be decreased if the subclavian artery on that side arises at or below the coarctation. Systolic blood pressure in the upper extremities is higher than in the lower extremities, but marked hypertension is uncommon. Pulse pressure in the lower extremities is narrowed, often 10 to 15 mm Hg. S3 is often present, and there may be an apical systolic ejection click (∼ half have a bicuspid aortic valve). A systolic ejection murmur from the coarctation may be heard at the left interscapular area over the back, and at the left upper sternal border. In the neonate a continuous murmur, when present, is usually from a left-to-right shunt across the ductus arteriosus, as collateral vessel flow is generally not audible until older age. Manifestations of congestive heart failure are those of combined left and right heart failure.
Chest radiograph can be nearly normal, but usually shows cardiac enlargement and pulmonary venous congestion.
ECG can be normal, but usually reveals right ventricular hypertrophy in the early months and left ventricular hypertrophy later.
Echocardiographic visualization of the aortic arch usually shows the site, length, and severity of coarctation and the aortic arch branching pattern. There is characteristically a constriction from the outer posterior curvature of the aortic wall, and an anterior peri-ductal shelf may be identified. An instantaneous systolic gradient may be derived from the velocities across the coarctation but may underestimate the severity of the lesion if cardiac output is depressed. The descending aortic flow has a characteristically diminished systolic upstroke velocity and prolonged antegrade flow.
Magnetic resonance and CT imaging can be used to delineate anatomic features not evident with echocardiography (Fig. 33-35).
Complex Coarctation and Aortic Interruption
Infants with severe isolated and complex coarctation usually present with congestive heart failure in the early neonatal period. Generally, the younger the infant, the more severe and complex are the combined malformations. Complete interruption of the aortic arch is usually associated with a ventricular septal defect and a systemic patent ductus arteriosus and is clinically indistinguishable from complicated coarctation. It is frequently seen as part of DiGeorge syndrome, which may have additional manifestations of hypocalcemia, absent thymic shadow on the initial chest radiograph, and possible impaired immune response to transfused viable nonirradiated leukocytes. Besides the findings described for simple coarctation, there is evidence of a large left-to-right shunt and pulmonary artery hypertension. Femoral pulsations may wax and wane, depending on ductal
caliber. A pan-systolic murmur of a septal defect or mitral regurgitation may be found. Ductal closure may result in a critically ill baby with poor perfusion, metabolic acidosis, and possibly disseminated intravascular coagulation, necrotizing enterocolitis, and renal and hepatic dysfunction.
caliber. A pan-systolic murmur of a septal defect or mitral regurgitation may be found. Ductal closure may result in a critically ill baby with poor perfusion, metabolic acidosis, and possibly disseminated intravascular coagulation, necrotizing enterocolitis, and renal and hepatic dysfunction.
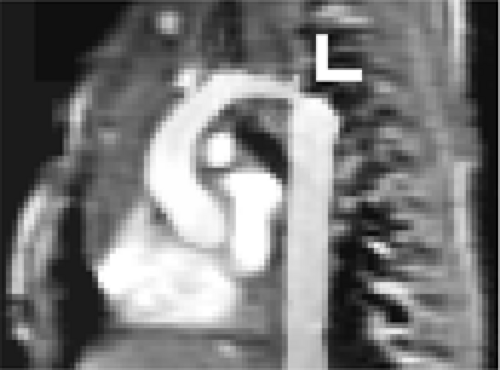 Figure 33-35 Magnetic resonance image of aortic coarctation in a 1 day old. Arrow indicates focal coarctation in the distal end of a hypoplastic aortic arch. |
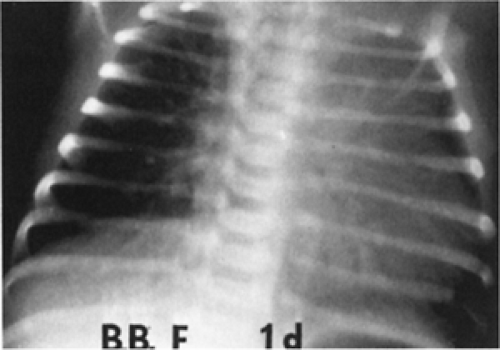 Figure 33-36 A chest radiograph of a 1-day-old infant with complex coarctation of the aorta shows marked cardiac enlargement and pulmonary vascular engorgement. |
Chest radiographs show considerable cardiac enlargement, pulmonary plethora, and edema (Fig. 33-36).
ECG can be normal, but often has right axis deviation, right atrial hypertrophy, right ventricular hypertrophy, and sometimes diminished left ventricular forces.
Echocardiographic examination delineates the aortic arch anatomy, and reveals associated lesions, including mitral and aortic stenosis, ventricular septal defect, sub-aortic obstruction, and conotruncal abnormalities.
Differential Diagnosis
Aortic arch obstruction should be suspected in any critically ill term baby with a septic-like shock. It should also be suspected as an associated anomaly in young babies with intracardiac anomalies such as ventricular septal defect, single ventricle, truncus arteriosus, and aortic or mitral valve disease who develop signs of poor systemic output. A thorough examination, with careful pulse palpation and blood pressure measurement in all 4 extremities, should lead to the correct diagnosis (see Tables 33-14 and 33-15). Infants presenting before 1 month of age usually have severe or complex coarctation. The presence of a ductus arteriosus supplying the descending aorta may be demonstrated by the finding of a lower arterial PO2 in the legs than in the arms. The hypoplastic left heart syndrome produces a similar shock-like picture or congestive failure in the first week as the ductus arteriosus closes. In these patients, there is cyanosis (See Color Plate), the peripheral pulses are diminished throughout, and the ECG shows marked diminution in left ventricular forces.
Treatment
All neonates with congestive heart failure thought to have coarctation of the aorta should be promptly hospitalized, treated, and examined by echocardiography. Infants with complex coarctation and aortic interruption become symptomatic because of constriction of the ductus arteriosus. Prostaglandin E1 infusion can dilate the ductus, restore systemic perfusion, improve metabolic abnormalities, and support life during the time needed to study the anatomy and arrange for surgery. Inotropic support with intravenous dopamine or adrenergic agents is often needed. In critically ill babies, there may be adverse ischemic consequences for the gastrointestinal, renal, hepatic, and coagulation systems. Echocardiographic examination usually provides the anatomic detail needed for surgery. If needed, cardiac catheterization, digital subtraction angiography, or magnetic resonance imaging may be useful for additional delineation of the aortic arch and intracardiac anatomy.
If after initial clinical management to effect stabilization, improvement or deterioration occurs, surgery should not be unduly delayed. The surgical procedures employed depend on the severity of the lesion and include resection of the coarctation with primary anastomosis, subclavian or prosthetic patch aortoplasty, or construction of a conduit from ascending to descending aorta; division of the patent ductus arteriosus; and, if needed, intracardiac repair of additional defects such as a large ventricular septal defect. The mortality rate for infants with complicated coarctation is 85% without surgery. Surgery increases the survival rate to 85%. Regardless of the type of coarctation, the mortality is related to age of presentation and is higher for those with duct-dependent descending aortic flow. In some infants with simple and milder coarctation who respond well to medical therapy, surgery may be delayed. Those who undergo surgical coarctation repair early in infancy may develop re-stenosis later, which may require re-operation or catheter balloon dilation. The survivors need close medical supervision throughout childhood and may require other operations for various associated abnormalities later. Catheter balloon dilation of un-operated primary discrete coarctation can offer palliation in the complex critically ill infant, but only in selected infants with an otherwise good size aortic arch does it appear to it provide long-lasting relief (50).
Aortic Stenosis
Fusion of the right-left or right-non commissures of the aortic valve, resulting in a bicommissural, functional “bicuspid aortic valve, is one of the most common congenital anomalies, occurring in 1.5% of the population. The resulting valve orifice may be diminished, but the effective stenosis is usually mild in neonates. Only very severe aortic valve narrowing produces symptoms or requires intervention in early infancy (Fig. 33-37). Critical isolated aortic valve stenosis is rare, and usually the result of fusion of both the right-non and right-left commissures, with the valve orifice too small to allow adequate cardiac output at physiologically obtainable left ventricular pressures. In this situation, adequate systemic blood flow (before relief of the aortic stenosis) is dependent on a patent ductus arteriosus that
allows right-to-left blood flow from the pulmonary artery into the aorta.
allows right-to-left blood flow from the pulmonary artery into the aorta.
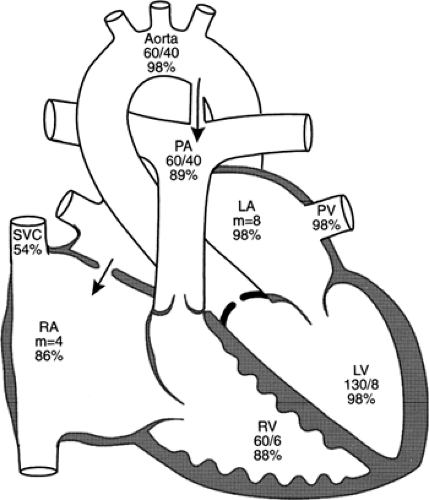 Figure 33-37 Diagram of the cardiac anatomy and physiology in a 1-month-old infant with valvar aortic stenosis had a systolic pressure gradient of 70 mmHg across the aortic valve. The blood passing from left to right through the ductus must return again through the aortic valve, with the excess flow compounding the obstruction. The large atrial shunt, whether a true anomaly or a sprung foramen ovale, elevates left atrial pressure. The numbers below the chamber name are pressure measurements in mmHg determined at cardiac catheterization; the percentages indicate oxygen saturation data. LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RA, right atrium; RV, right ventricle; SVC, superior vena cava. Adapted from Mullins CE, Mayer DC. Congenital heart disease: a diagramatic atlas. New York: Alan R Liss, 1988, with permission. |
The symptoms are those of congestive heart failure, pulmonary edema, and sometimes peripheral vascular collapse. The baby may appear ashen and cyanotic if the pulmonary edema is severe. The cardinal features are tachypnea, a blowing systolic murmur at the upper right or middle left sternal border and an apical early systolic click resembling a split S1. If the valve orifice is very small, forward valve flow is small, and the murmur may be soft.
Chest radiographs show cardiac enlargement and pulmonary venous congestion.
ECG usually has biventricular hypertrophy with T-wave changes (see Table 33-14).
Echocardiographic examination demonstrates a deformed immobile aortic valve with commissural fusion. In severe aortic obstruction, trans-valvar flow is diminished, and the systolic murmur and the Doppler-derived pressure gradient are of low amplitude and do not reflect the severity of the lesion. The left ventricle appears hypertrophied and may have decreased or dilated internal dimensions and poor or hyperdynamic systolic function. Some patients may have coarctation and mitral valve abnormalities. Echocardiography can also identify those neonates with associated hypoplasia of the left ventricular chamber, mitral and aortic annuli and aortic root that do not benefit adequately from valvuloplasty and require a staged hypoplastic left-heart-type surgical approach for survival (51).
Treatment of critical stenosis consists of stabilization with administration of inotropic support, oxygen, and frequently prostaglandin E1 to allow right ventricular support to the systemic circulation; to be followed as soon as feasible by balloon valvuloplasty or surgical valvotomy (52). This provides effective palliation in infancy. Although many require repeat valvuloplasty during childhood, this is usually accomplished with success. Ultimately, and hopefully after childhood growth, many of the worse valves will require surgical replacement with a prosthetic valve or pulmonary autograft (Ross procedure). The asymptomatic infant with auscultatory findings of aortic stenosis and those after valvuloplasty require serial evaluation because, over the long term, valvar aortic stenosis very frequently progresses and recurs to some degree.
Acyanotic Anomalies with Left-to-Right Shunt
Ventricular Septal Defect
Ventricular septal defects may be small or large, single or multiple, and isolated or associated with other cardiovascular malformations. They are an integral part of complex congenital heart disease lesions, such as tetralogy of Fallot, truncus arteriosus, double-outlet right ventricle, and atrioventricular canal, and they have been associated with virtually every other known congenital cardiac malformation. Small, isolated self-closing muscular ventricular septal defects, frequently detectable only by echocardiography, are the most common congenital cardiac anomaly, occurring in 2% to 3% of term newborns (2) and more frequently in infants born prematurely (see Tables 33-1 and 33-2). Large defects occur most commonly singly in the membranous septum, less often in the low portion of the muscular septum, infrequently beneath the pulmonary valve, or posterior next to the tricuspid valve. Even though only 10% of ventricular septal defects cause symptoms, they remain the most common cause of congestive heart failure after the second week of life (see Tables 33-7). Although small defects are usually isolated anomalies, extra-cardiac malformations occur in up to 24% of the patients with large defects. Recognition of a large ventricular septal defect remains paramount because without closure a large defect with pulmonary hypertension can lead to irreversible Eisenmenger-type pulmonary vascular disease by as early as the first birthday.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


