Fetal lateral cerebral ventriculomegaly (arrows). (a) Mild ventriculomegaly; (b) unilateral mild ventriculomegaly; (c) severe ventriculomegaly.
A value of >15 mm indicates severe ventriculomegaly. This is almost always associated with an intracranial malformation, although the outcome is variable and depends largely upon the underlying etiology of the ventricular dilatation. The available studies suggest that fetuses with isolated severe ventriculomegaly have an increased risk of perinatal death and a probability of severe long-term neurologic sequelae in approximately 50% of survivors[2].
An intermediate value of the atrial width, 10–15 mm, is commonly referred to as mild ventriculomegaly and is less frequently associated with many CNS anomalies, including agenesis of the corpus callosum and open neural tube defects[2,3]. Mild ventriculomegaly is associated with chromosomal aberrations: the risk of trisomy 21 is increased 3.8 times when ventricular enlargement is an isolated finding[4]. Fetal infections, such as cytomegalovirus, may result in ventricular enlargement, although usually with other sonographic abnormalities (cerebral echogenicities, microcephaly and porencephaly). When associated anomalies are ruled out, most infants are completely asymptomatic after birth. However, several reports have indicated that some fetuses develop severe cerebral anomalies in advanced gestation or after birth (hydrocephalus, white matter injury and cortical plate abnormalities) and have suggested an increased risk of neurologic compromise. Studies that have investigated the prognosis of mild ventriculomegaly have been limited by the lack of standardized follow-up protocols and difficulty in defining ‘normality.’ However, most fetuses develop normally, and only about 10% demonstrate neurodevelopmental abnormalities of variable types and magnitude[3].
It is commonly accepted that when mild ventriculomegaly is encountered, all efforts must be made to rule out associated anomalies. A detailed expert sonographic survey of fetal anatomy is mandatory and invasive testing for cytogenetic analysis should be considered. Detailed ultrasound assessment may necessitate scanning by the vaginal route. There is debate as to whether fetal magnetic resonance imaging (MRI) is indicated in all cases of apparently isolated ventriculomegaly[3]. This technique may provide significant diagnostic information, particularly in the last trimester of gestation. In continuing pregnancies, there is no indication to modify the standard obstetric management.
Key counseling points
(1) Offer an expert sonography survey of fetal anatomy, karyotype and serial scans to assess progression.
(2) Consider a maternal infection screen (cytomegalovirus, toxoplasomsis).
(3) Consider fetal MRI to exclude subtle intracranial pathology, including neuronal migration disorders and intracranial bleeding.
Neural tube defects
Anencephaly is characterized by the absence of the cranial vault and telencephalon. The diagnosis is easy in the second and third trimester, and relies upon the demonstration of the absence of the cranial vault. However, most cases can be confidently identified at the 11–13 weeks’ scan. At this time, a cephalic pole is usually present, but appears overtly abnormal because of the lack of an ossified calvarium[5] (Figure 6.2). The terms acrania and excencephaly have also been used to describe such an appearance, which represents an early stage in the development of anencephaly. The outcome is uniformly fatal.
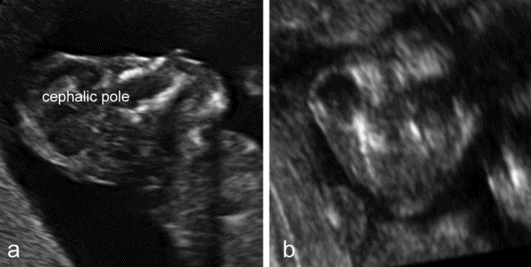
Anencephaly. (a) At 12 weeks’ gestation, a cephalic pole, albeit abnormal, is visible. (b) From midgestation, the cephalic pole is usually completely absent.
Spina bifida is commonly subdivided into open and closed forms. Open spina bifida is characterized with a full-thickness defect of the skin, underlying soft tissues and vertebral arches exposing the neural canal. The defect may vary considerably in size. The lumbar, thoracolumbar or sacrolumbar areas are most frequently affected. Leakage of cerebrospinal fluid through the defect causes an increased concentration of alpha-fetoprotein and acetylcholinesterase in the amniotic fluid and maternal serum, but these are no longer used for diagnostic purposes. Open spina bifida can be identified sonographically by demonstrating the opening of the neural tube and the defect of the overlying soft tissues. A cyst formed by the fusion of the malformed cord and meninges (myelomeningocele) is usually found. In a minority of cases, there is no covering membrane (myelocele). The diagnosis of a neural tube defect may be difficult and always requires meticulous scanning. Examination of the fetal head is useful, as open spina bifida is consistently associated with easily recognizable cranial lesions. Leakage of cerebrospinal fluid leads to displacement of the cerebellum and medulla oblongata through the foramen magnum inside the upper cervical canal (Chiari type II or Arnold–Chiari malformation). Sonographically, this results in small head measurements at midgestation, obliteration of the cisterna magna, small size and abnormal shape of the cerebellum that is impacted deep into the posterior fossa (banana sign), frontal bossing (lemon sign) and ventriculomegaly. The detection rate of a midtrimester scan is in the range of 90%[6,7]. Abnormalities of the posterior fossa and lateral ventricles have also been described at the 11–13 weeks’ scan, but the diagnostic accuracy of these findings to screen for spina bifida is still being assessed.
There is a correlation between the site and extension of the spinal lesion and the neurologic outcome. Indeed, the lower and smaller the defect, the less severe the neurologic compromise. Fetal medicine experts often record the level of the lesion by counting the affected vertebrae either from the most distal one, the fourth sacral vertebra (S4), or from the 12th thoracic vertebra in the rib cage (T12). Antenatal ultrasound assessments correlate reasonably well with postnatal assessment (75%) and MRI adds little in this respect, but it is impossible to predict with precision motor functions, morbidity and developmental milestones[8]. On the other hand, ventricular enlargement does correlate with the need for postnatal shunting[9]. Antenatal counseling is complex and should involve a multidisciplinary team, including pediatric neurosurgeons. Postnatal hydrocephalus requiring shunting and incontinence, and motor weakness requiring wheelchair support are common. Most women will chose termination of pregnancy, although an increasing number may choose expectant management and even contemplate intrauterine treatment (see Chapter 28). It has been suggested that cesarean section may ameliorate the neurologic outcome of infants with open spina bifida, but the evidence is weak. As a rule, a cesarean section is only performed for obstetric indications.
Closed spina bifida is characterized by a vertebral schisis covered by skin. Most defects are small, involving only few veretebral segments, and the classical intracranial signs (lemon-shaped skull, banana-shaped cerebellum) are always absent. As a consequence of this, diagnosis with sonography is difficult and, in practice, is only possible in those cases associated with a subcutaneous mass, a meningocele or lipoma, overlying the defect[6]. The outcome of closed spina bifida is difficult to predict. These infants do not develop Arnold–Chiari malformation and hydrocephalus. Particularly those with subcutaneous masses, however, may suffer from neurologic sequelae of variable entity, including weakness or paralysis of the legs and incontinence, which are usually a consequence of tethering or compression of the spinal cord.
The term cephalocele indicates a protrusion of intracranial contents through a bony defect of the skull. In most cases, the lesion arises from the midline, in the occipital area, less frequently from the parietal or frontal bones. Encephaloceles are characterized by the presence of brain tissue inside the lesion. When only meninges protrude, the term cranial meningocele is used. Cephaloceles often cause impaired cerebrospinal fluid circulation and hydrocephalus, and massive encephaloceles may be associated with microcephaly. Associated anomalies are frequent. Fetal cephaloceles are suspected when a paracranial mass is observed on sonography. The diagnosis of encephalocele is easy, as the presence of brain tissue inside the sac is striking on ultrasound. Differentiation of a cranial meningocele from soft tissue edema or a cystic hygroma of the neck may be difficult. The diagnosis of cephalocele is favored when it is possible to demonstrate a defect in the cranial vault or cerebral abnormalities, such as ventriculomegaly. The pediatric literature suggests that the outcome of cephaloceles is mainly related to the presence or absence of brain tissue inside the lesion. However, the largest available antenatal series reports a dismal prognosis for both varieties[10], with the possible exception of small lesions in the presence of a normal intracranial anatomy (Figure 6.3).
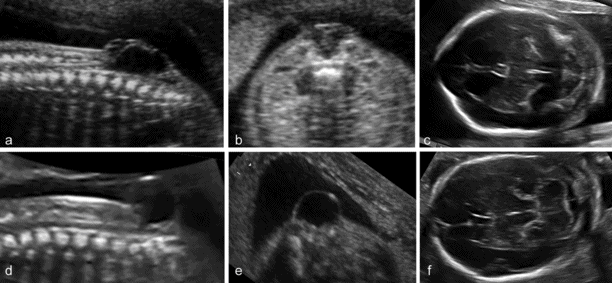
(a–c) Open spina bifida (myelomeingocele): large vertebral defect with an overlying septated cyst associated with typical findings of Arnold–Chiari malformation, cranial bossing and obliteration of the cisterna magna. (d–f) Closed or skin-covered cystic spina bifida (meningocele): a small vertebral defect with an overlying nonseptated cyst, and normal intracranial anatomy.
Key counseling points
(1) Fetal karyotyping is not indicated in apparently isolated neural tube defects.
(2) With open spina bifida, correlation between the level of the defect and postnatal function is relatively poor.
(3) Closed spina bifida has a better outcome than open spina bifida, but neurologic compromise is possible and is difficult to predict.
(4) Large cephaloceles, particularly in the presence of abnormal intracranial anatomy, have a poor outcome.
Midline anomalies
The holoprosencephalies are complex abnormalities of the forebrain that share in common an incomplete separation of the cerebral hemispheres and formation of diencephalic structures. They are rarely seen at birth. However, they have a high intrauterine fatality rate and are a relatively common group in the series of antenatally detected CNS anomalies. The etiology is heterogeneous. In most cases, the anomaly is isolated and sporadic. In other cases, chromosomal abnormalities (trisomy 13 and polyploidy) and/or anatomic abnormalities are found.
The most widely accepted classification of these disorders recognizes three major varieties: the alobar, semilobar and lobar types. More recently, middle interhemispheric variant has been added. The alobar, semilobar and middle interhemispheric variant types have a similar appearance on antenatal ultrasound – there is no midline echo anteriorly and there is a single ventricular cavity (Figure 6.4). Gross facial abnormalities (cyclopia, hypotelorism and absence of the nose) are frequent. In most cases the diagnosis is possible at the 11–13 weeks’ scan. The lobar variety is associated with more subtle findings (absence of the septum pellucidum with central fusion of the frontal horns) and the diagnosis is rarely made prior to 20 weeks’ gestation[11].
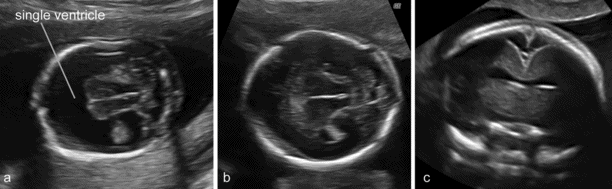
(a) A typical case of fetal holoprosencephaly: a large single ventricular cavity is seen and there is no midline in the anterior part of the brain. (b) A case with less obvious findings. (c) The anterior midline is absent but the single ventricular cavity can only be clearly demonstrated with a transvaginal coronal scan.
The invariably poor prognosis for infants affected by a lobar and semilobar holoprosencephaly is well established. Termination of pregnancy should be offered. Even cases with antenatally diagnosed lobar holoprosencephaly have an extremely poor postnatal neurodevelopment.
Agenesis of the corpus callosum (ACC) is an anomaly of uncertain prevalence and clinical significance. The best estimates suggest prevalence of around 1.4 per 10,000 live births in the general population and 2–3% in the developmentally disabled. The etiology is heterogeneous. Genetic causes are predominant, and the anomaly is frequently a part of many different syndromes. The high frequency of associated malformations and chromosomal aberrations suggest that ACC is often part of a widespread developmental disturbance. ACC may be either complete or partial. In the latter case, also referred to as dysgenesis, the caudad portion is missing to varying degrees. Hypoplasia refers to a corpus callosum (CC) of normal length but with much decreased thickness, and is rarely identified prenatally.
Although the anatomy of complete ACC is variable, this condition is usually suspected in routine examinations at midgestation because of the absence of the cavum septi pellucidi (CSP) and the presence of a peculiar configuration of the lateral ventricles characterized by an increased separation of the frontal horns and a mild enlargement of the posterior horns (tear-drop ventricles), frequently associated with mild ventriculomegaly. The definitive diagnosis is made by demonstrating the absence of the CC in coronal and/or sagittal scans (Figure 6.5)[12]. Diagnosis of partial agenesis is also possible, but the sonographic findings are more elusive than with the complete form. The CSP is usually present and the axial sections may be completely unremarkable. The diagnosis requires a sagittal section demonstrating that the CC shorter than normal and does not form a complete arch over the third ventricle (Figure 6.6). Normal charts of the size of the CC are available, but should be used with caution because partial agenesis is a rare condition and it has not been possible thus far to establish a clear threshold for pathologic appearance. Most cases diagnosed antenatally have shown a reduction in size of 50% or more. We have seen many cases with measurements <5th centile and, as expected, most resulted in the birth of normal children (Figures 6.5 and 6.6).
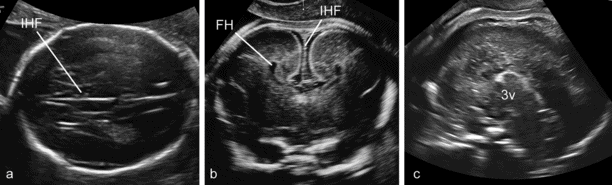
Complete agenesis of the corpus callosum (CC). (a) The axial plane reveals mild enlargement of the lateral ventricles that have a typical tear-drop shape, absence of the cavum septi pellucidi (CSP) and distension of the interhemispheric fissure (IHF) that is centrally partitioned by the falx cerebrii. (b) The coronal plane demonstrates the large IHF, the absence of any bridging structure connecting the two hemispheres, and the lateral separation of the frontal horns that appear medially concave. (c) The sagittal plane demonstrates the absence of complex formed by CC and CSP over the third ventricle. FH, frontal horns.
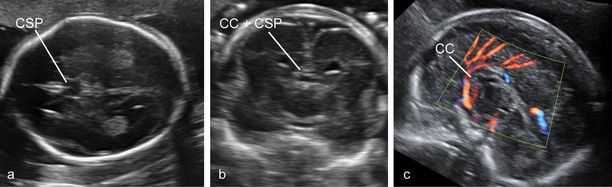
A panel of images highlighting the difficulties in the diagnosis of partial agenesis of the corpus callosum. (a) The axial plane reveals a rather normal-looking cavum septi pellucidi (CSP). (b) The coronal plane demonstrates the presence of the complex formed by the corpus callosum (CC) and CSP bridging between the two hemispheres. (c) Only the sagittal plane demonstrates a CC that is at the same time quite short (the arch does not cover entirely the area of the third ventricle, and the measurement is about 50% the expected size at this gestational age) and irregular in shape.
ACC, be it complete or partial, is frequently a part of syndromes or multiple malformations. A recent systematic review assessed the rate of neurodevelopmental outcome in 132 fetuses (16 studies) with isolated ACC[13]. The authors reported neurodevelopmental outcome as: normal; borderline or moderate disability; or severe disability. In complete ACC, the respective figures were 74.3%, 14.3% and 11.4%. The outcome was slightly less favorable, although not significantly different, for partial ACC (65.5%, 6.9% and 27.6%, respectively). When taking into account only those studies using MRI and standardized tools of neurodevelopmental assessment, in complete ACC, the rates were 83.7%, 8.2% and 8.2% for normal, borderline/moderate and severe disability, respectively. The corresponding rates for partial ACC were not reported due to the small number of cases. The authors of the review highlighted many limitations in existing studies, including limited and inconsistent data that prevented subgroup analyses. Providing a precise estimate of the risk of neurodevelopmental delay is difficult. One important limitation of the available studies is the time of follow-up. In most cases, assessment was made in the preschool period. This may represent an important shortcoming as in one series a progressive decline of intellect was noted, with a considerable number of infants demonstrating learning difficulties in school.
The septum pellucidum, with the inferior fornix, forms the medial border of the lateral ventricles. It contains two leaves that in fetal life are separated by a fluid-filled cavity, the CSP. The leaves fuse in late gestation or immediately after birth and the cavity is seen only in a minority of adults. Absence of the septum pellucidum in fetuses is frequently associated with a number of cerebral anomalies, including ACC, holoprosencephaly, destruction secondary to raised intraventricular pressure, schizencephaly and septo-optic dysplasia (deMorsier syndrome). Isolated absence of the septal leaves with central fusion of the frontal horns is possible, and is usually of no consequence (Figure 6.7). The greatest challenge is to differentiate isolated absence from septo-optic dysplasia. Visualization of the optic chiasma by either Magnetic resonance or three-dimensional ultrasound is useful, although it does not seem to allow a definitive diagnosis. The available experience is limited, but thus far, two-thirds of fetuses with absent CSP were reported to be normal at birth. If a normal optic chiasma can be demonstrated, the probability increases to 90%[14].
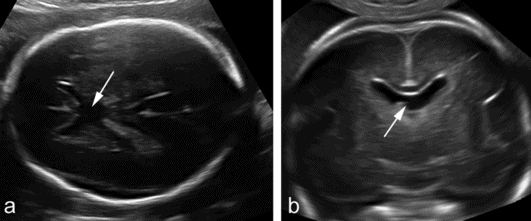
Absence of the septum pellucidum demonstrated in (a) an axial and (b) transvaginal coronal plane. The two frontal horns communicate centrally and there is no evidence of the leaves of the septum pellucidum (arrows).
Key counseling points
(1) Holoprosencephaly is frequently associated with trisomy 13 and, in general, has a very poor prognosis. The lobar type, although associated with an equally poor prognosis, may be difficult to diagnose antenatally.
(2) ACC is frequently a part of syndromes or multiple anomalies. Offer a detailed scan, MRI and karyotype, including microarray testing. There is no need, however, to test for infections.
(3) Three out of four children with complete isolated ACC will have normal neurodevelopmental outcome, at least at pre-school age; partial agenesis may represent a different clinical entity than complete agenesis, but the available data are limited.
Cystic and cyst-like abnormalities of the posterior fossa
Abnormal fluid collections in the fetal posterior fossa encompass a wide spectrum of different entities, ranging from normal variants to severe anomalies (Table 6.1).
In the late first trimester, the fourth ventricle is large and a relatively small cerebellum is found on top of it. In the following weeks, the cerebellum grows to enfold completely the fourth ventricle. However, a small finger-like appendage of the fourth ventricle, the Blake’s pouch, is frequently seen protruding into the cisterna magna, caudally to the cerebellum. It has been suggested that there is a continuum of anatomic anomalies involving the fourth ventricle–Blake’s pouch complex (Figure 6.8)[15]. The mildest of these anomalies is the Blake’s pouch cyst, an isolated persistence of the Blake’s pouch. This term was originally introduced in pediatric neuroradiology to indicate a compressive cyst of the posterior fossa displacing superiorly the cerebellar vermis and causing obstructive hydrocephalus. More recently, the same term has become popular in fetal imaging studies to indicate cases with a posterior fossa cyst displacing superiorly an intact cerebellar vermis, typically in association with a normal ventricular system and a normal size of the posterior fossa. The entity described in the original neonatal studies and the one later described in fetal studies are likely to be different, as the latter has typically a normal outcome and appears to be rarely associated with ventriculomegaly. Megacisterna magna may be a variation of the Blake’ pouch cyst that does not result in superior displacement of the vermis.
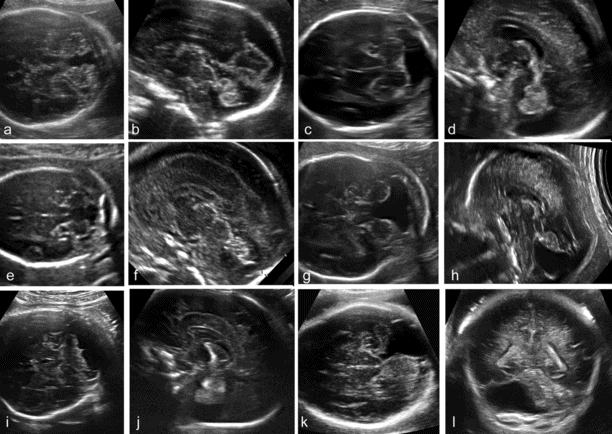
Categorization of posterior fossa fluid collections (see Table 6.1). (a, b) Blake’s pouch cyst; (c, d) megacisterna magna; (e,f) vermian hypoplasia; (g, h) Dandy–Walker malformation; (i, j) cerebellar hypoplasia; (k, l) arachnoid cyst of the posterior fossa.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


