The heart examination is a critical moment in fetal ultrasound (US). After birth, cardiologists carry out this examination, but before then the fetal heart is seen by non-cardiologists. While it is not necessary to have the level of knowledge of a pediatric cardiologist to perform this systematic prenatal check-up, it is essential to acquire a simple, solid knowledge base if we are to carry out fetal heart examinations that are valid and have a long-term prognostic value.1
We hope that this will be the sort of book that we would have wanted to find years ago when we began studying fetal hearts. There are now excellent “classical” reference books that teach fetal cardiology. They usually contain much more than what is needed in our daily practice, and because of this, require long and attentive research to find the answers essential to fetal US. What we are trying to do here is provide a practical guide for the US practitioner, underlining the elements that we have found to be essential based on our experience.
The three of us are practicing fetal US specialists, each with different but constantly evolving and complementary interests and skills. Claudio Lombardi joined our team for this new edition because of his expertise in first trimester examination.2 Jean-Eric Develay–Morice is still involved in the technical aspects of our craft, specifically concentrating on finding new ways to diagnose previously “undiagnosable” pathologies. I spent many years in the anatomic examination of thousands of normal and pathological fetal hearts, using a strict segmental analysis. I apply this work to US–anatomic correlations. Over the years of close collaboration, we found that, above all, the US specialist needed tools to test for what we call “normality”.
Our experience has shown that the pathologies involved in the fetuses with the worst prognosis were always of the same type. We learned that what is “essential” is to be able to say that a fetal heart looks normal by checking for simple warning signs, rather than being able to precisely diagnose all types of pathologies. This is seen to be true in a great majority of cases.
When faced with a cardiopathy, the first role of the practitioner is to ensure that there is not an associated extracardiac pathology.
The warning signs we propose are simple, and we explain how to check for them. We do this using visual comparisons, which come from our experience gained during teaching numerous workshops in France and the Mediterranean region. We also kept what worked best in our original French edition, the same style, and the same conscious insistence on repetition.
Repetition has proved to be an essential part of our methodology, providing a book that is fundamental and practical for daily use. To increase its value as a training and reference tool, we have added references and clips which include US sequences, a large section on fetal heart anatomy, and anatomic and US correlations. We hope that you will find in this book and its clips practical keys to the practice of fetal heart US.
One of our primary intentions is to introduce information that is indispensable for verifying normal fetal heart architecture, as well as information useful in the detection of important pathologies. The precise diagnosis of cardiopathies, and their prognoses, remains the realm of the pediatric cardiologist.
The US practitioner has several aims. One is to verify normal fetal cardiac architecture, which also involves looking for cardiopathies in the case where another anomaly has presented itself during morphological examination. The discovery of an isolated cardiopathy is a rare event.
Here we will lay out a simple methodology capable of verifying normal fetal cardiac architecture in 3 steps and 10 key points. These key points have been defined through a series of US–anatomic correlations and tested by numerous US specialists. The diagnostic criteria are easily accessible and allow us to eliminate the cardiopathies as defined by an expert consensus.3
We begin by reviewing the knowledge essential in understanding what is normal, as well as pathologic, in the fetal heart. We then touch on those physical principles crucial in optimizing the examination itself, finally introducing our methodology. Next, we study the pathologies themselves, outlining the pitfalls involved in studying each one while proposing the best methods for avoiding these traps. Finally, we will describe the type of morphological examination necessary in those cases where a cardiopathy has been discovered, concluding with a review of points to remember.
General notions
In the fetus there are two types of cardiopathies that are important to detect:
In this category of cardiopathies, you must equally eliminate the possibility of:
When faced with certain cardiopathies whose prognosis is very bleak, or when the cardiopathy itself is a warning sign of a more complex pathology, the pediatric cardiologist might be led to propose a medical termination of pregnancy (MTP). Fetal pathologic verification is therefore highly recommended.5 With the family’s agreement, the fetal pathologist searches for those markers that were not visible during US in order to determine if the cardiopathy can be classified as a genetic syndrome or association. The results of this research allow us to propose appropriate genetic counseling for future pregnancies.
Sonographic measurement of the nuchal translucency (NT) thickness at 11–13 weeks is a well-established screening method for aneuploidies and other major anomalies. Fetuses with increased nuchal translucency and normal karyotype have a high frequency of congenital heart disease (CHD).8,9 This justifies an early fetal heart examination.10
However, the main goal of first trimester scan will be to offer a systematic US investigation of fetal structures as a significant number of major anomalies (CHD included) are present in fetuses with normal NT.
In pregnancies without particular problems, the heart is systematically verified by morphological US at 20–22 weeks. Even when considered normal at this stage, the heart should be observed by growth US at 32 weeks. It seems clear that a strictly architectural pathology could not have begun to form between the 22nd and 32nd gestational weeks. For instance, a heart found to have an AVSD at a gestational age of 32 weeks was not normal earlier at 22 weeks; AVSD was present during the very first weeks of development. On the other hand, certain pathologies, even architectural ones, evolve in relation to flow. Pathologies such as pulmonary stenosis, which were invisible or poorly visualized, even passing unseen at 22 gestational weeks, can be individually observed at 32 weeks.
The result of such screening on the detection of CHD has been studied in Europe,11 showing variations depending on the different methods and protocols in use. The countries having the best results are those where every woman has access to three US examinations (at 12 weeks for NT, around the 22nd week with a morphological examination, and the third examination at 32 gestational weeks to check growth and re-verify the morphology). In certain countries, where legislation has modified this practice, the first trimester examination is now of first importance12 and give significant results13 when practised as explained in Chapter 5 of this book.
Though rarely seen, the predictive value of cardiac anomalies is an extremely important warning sign of other anomalies (when considering all CHD taken together, and in relation to the increased risk of chromosomal abnormalities). Because of these observations, when a cardiopathy is discovered using US, the practice of determining the karyotype becomes indispensable.14 This is especially true because we now know that there are chromosomal anomalies in more than 33% of the cases studied (more than 15% in those cases where the cardiopathy appears isolated, and around 40% in those cases reported when the morphological examination reveals an associated anomaly).15 A complementary examination researching the deletion of chromosome 22q11 will be requested in the case of conotruncal malformations.16 Here we see the important role the fetal pathologic examination plays when a pregnancy has been medically terminated for an “isolated” cardiopathy. Markers that were difficult or impossible to see during US can be present here, such as a dysmorphology or visceral situs anomalies. This will allow us to identify known or unknown polymalformation syndromes.17
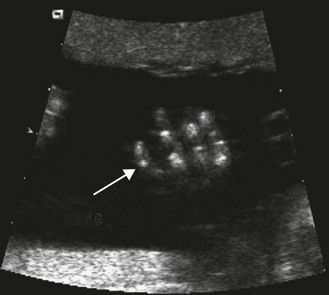
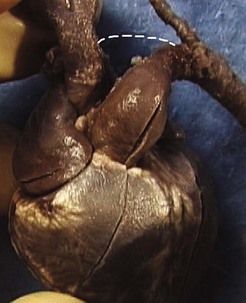
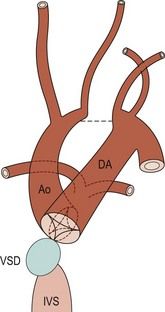
The frequency of karyotype anomalies also varies depending on the type of cardiopathy observed. We know that there is an occurrence of nearly 40% of trisomy 21 (T21) when an AVSD has been diagnosed.18 For instance, when faced with complete AVSD (Figs 1.1 and 1.2), if the karyotype is not known, we should first verify fetal nasal bone (NB).19 It is important to note that an incorrect size or absence (Fig. 1.3) here, like an amesophalangy or a brachymesophalangy (Fig. 1.4), represents an important complementary element. It is equally important to remember that in cases of IAA and coarctation of the B2 type (Figs 1.5 and 1.6),20 which is a rare form of CTC, over 80% show a microdeletion in 22q11. Note also that in this particular CTC, provoked by a neural crest pathology,21 the presence of an outlet VSD is constant (Fig. 1.7).
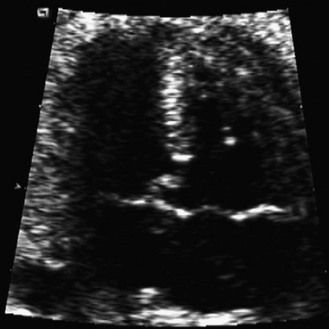
Figure 1.1 Apical four-chamber view. Complete AVSD associating an ASD ostium primum with LIAVV and inlet VSD.
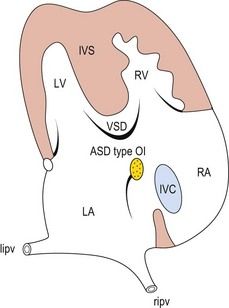
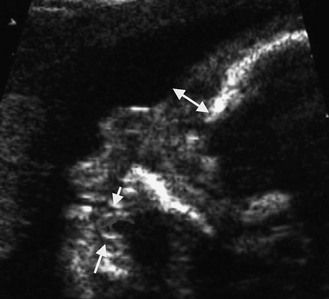
Figure 1.3 Ultrasound profile of a T21 fetus; NB absent. Note the lingual protrusion and the width of the prefrontal panicle (arrows).
Attention given to these confirmed anatomic findings in fetal pathology allow us to be sensitive to the presence of these pathologies, guiding our investigations. We know that:
The key to understanding our methodology is that it is based on the verification of the criteria for normality allowing us to eliminate any suspicion of important pathologies, rather than a strict sequential, segmental analysis,23 which creates an exhaustive search for each marker or each sign of any individual pathology. To use our method, we must clearly identify those elements that allow this verification, determining which views and images make this possible.
Criteria for normality
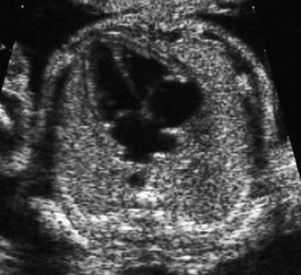
Figure 1.8 On this view of four normal chambers we see normal offsetting and visualization of two pulmonary veins. We do not find any form of the AVSD spectrum or TAPVR.
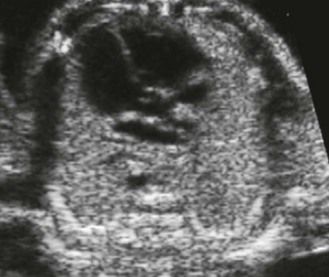
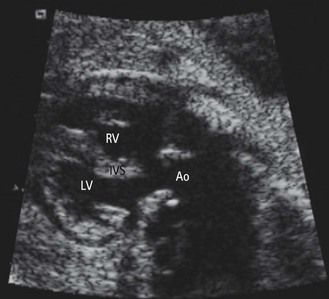
Figure 1.9 On this normal LV–Ao view we see a normal septal aortic continuity. There is no VSD by misalignment nor any atrioventricular discordance.
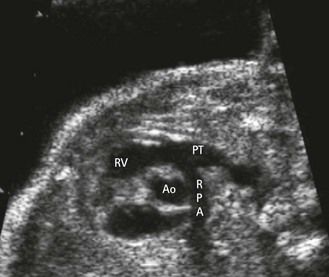
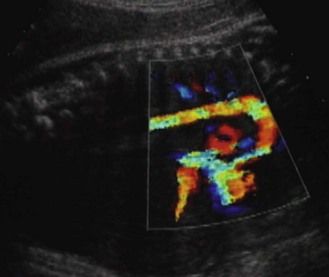
Figure 1.10 In this US view there is a normal ventricular–arterial concordance. There is no TGV or ventricular–arterial discordance.
Review
Simple points concerning the development, anatomy, and hemodynamic qualities of the fetal heart will improve our understanding and make the examination easier.
Development
While we know that simplistic explanations (we know to be “not really true”) can be of great help for the practitioner, we will not discuss here the recent and very complex findings about the heart development.27,28
The heart—whose architecture is definitive at a gestational age of 10 weeks in a fetus with a cranial rump length (CRL) of less than 4 cm—is the size of a grain of rice and beats at more than 160 times per minute.
Its evolution depends on genes of development and lateralization, primary and secondary heart fields as well as neural crests. The flow which begins to pass through it by the fifth week of gestation also contributes.
Seeing this in a very schematic way, the initial tube develops out of the islands of angioforming cells whose origin is splanchnopleuric and that are situated on the anterior pole of the embryo. Next, during the phase of delimitation, the explosive growth of the cephalic pole induces a coiling of the anterior pole of the embryo, causing the cardiac primordium to pivot by 180°. This then becomes ventral in relation to the cephalic pole and the stomodeum (the future mouth). The most anterior part of these tubes, formed by a symmetric tube pair that has become fused at their most distal section in the pericardiac cavity, becomes the cardiac tube itself. This tube then binds to the venous cardinal and vitellin systems (Fig. 1.12).
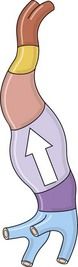
The intrapericardiac portion of the fused tube, due to its growth in a limited space, will normally form a right loop (Fig. 1.13). This will then develop into an organ that is both uneven and asymmetric.
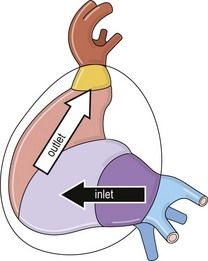
Figure 1.13 Left view of the tube after the formation of the right loop showing inlet and outlet flows.
The initial section, situated before the loop or inlet, is formed by a primitive atrium receiving the venous returns and communicating through a unique anulus, called the atrioventricular canal (AVC), with the primitive ventricle.
The distal section, situated after the loop, or outlet, is followed by the arterial system which is formed out of the common arterial trunk (CAT) and then followed by the aortic arches.
The evolution of this tube, with its chambers connected in series, is made by the compartmentalization of the inlet section. This section is located before the loop in a relatively orthogonal way in four chambers with:

Figure 1.14 Anterior view: vertical diagram of the heart before the division of the AVC and the initial CAT.
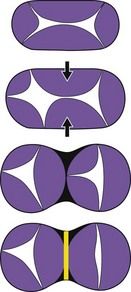
Figure 1.15 Anterior left view with a diagram of the stages of the transformation of the AVC into two annuli.
These phenomena, which are far more complex in reality, are still the focus of much fundamental research.28
We now arrive at the formation of parallel inlet tracts (Fig. 1.16), situated in the same plane.
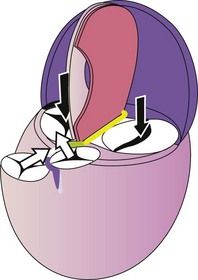
Figure 1.16 Left view diagram of the “verticalized” heart: septation achieved. Black parallel arrows represent the inlet flow; the orthogonal white arrows represent the outlet flow.
The outlet section undergoes far more complex modifications. Located after the loop, the distal section of the tube in which outflow is seen, is under the influence of neural crests.29 Here:
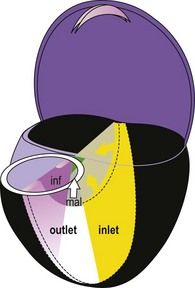
Figure 1.17 Diagram of the “verticalized” and opened heart. Localization of the inlet septum (yellow) and the outlet septum (alignment septum in white; infundibular in purple).
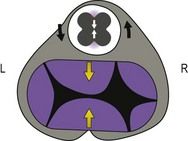
Figure 1.18 Posterior right view of the fetal heart after ablation of the atria and the great vessels, showing the initial CAT and initial AVC.
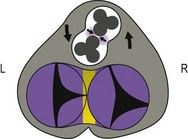
Figure 1.19 Intermediary stage. Evolution: partial AVC dominated by two vessels in the process of spiraling and wall development.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


