117 Trisomy 21
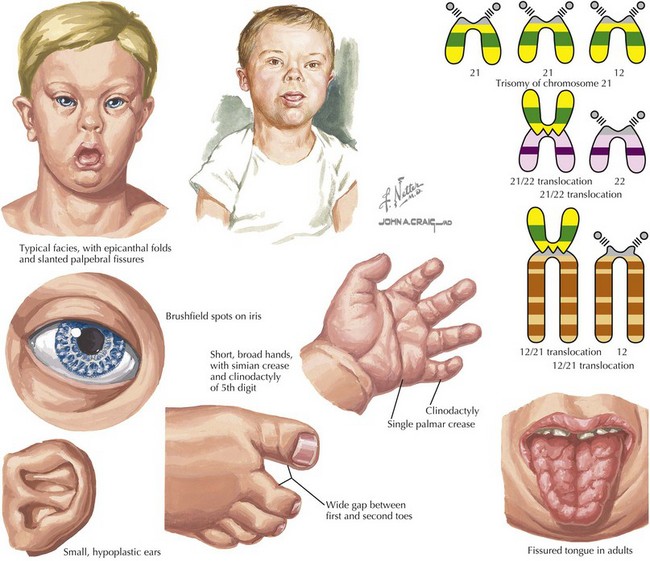
Down syndrome, or trisomy 21, is the most common chromosomal abnormality among live-born infants and is the most frequent microscopically identifiable genetic cause of mental retardation. An extra copy of chromosome 21 results in an overexpression of the genes found on this chromosome to result in the phenotypic differences seen in patients with Down syndrome. This disorder is characterized by a variety of dysmorphic features, congenital malformations, and medical problems (Figures 117-1).
Etiology and Pathogenesis
Although trisomy of chromosome 21 (e.g., 47,XY, +21) in which a complete extra copy of chromosome 21 is present, causes approximately 94% of cases, other causes are also seen. Approximately 2% to 3% of cases of trisomy 21 can present with mosaicism (e.g., 47,XY, +21 [50%] or 46,XY[50%]) in which there is a mixture of trisomic cells and normal cells. This can lead to an attenuated phenotype, but it depends on the relative mosaicism of different tissues. Finally, approximately 3% to 4% of cases are caused by a Robertsonian translocation, in which the q arm of chromosome 21 is translocated onto another chromosome. This results in a fetus with 46 chromosomes but three copies of the long arm of chromosome 21, which carries all of the functional genes of this chromosome (e.g., 46,XY,der(15 : 21)(q10;q10), +21; Figure 117-1). Although most Robertsonian translocations are new mutations and occur regardless of maternal age, it is important to distinguish the cause and rule out a translocation carrier parent, which substantially increases the risk of recurrence.
Clinical Presentation and Differential Diagnois
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


