CHAPTER 35 The genetics and molecular biology of gynaecological cancer
Introduction
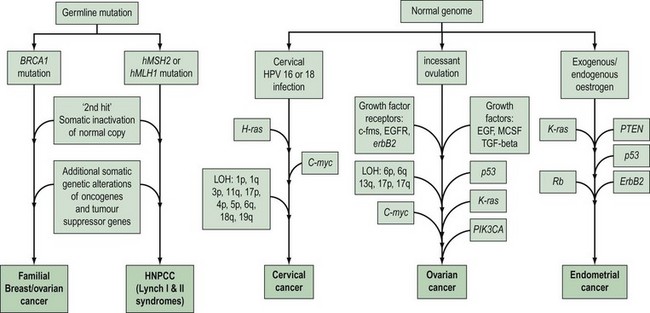
Gynaecological cancers provide examples of a number of contrasting mechanisms of carcinogenesis (Figure 35.1). For example, ovarian and endometrial cancer can occur as components of familial cancer syndromes (familial breast/ovarian cancer and Lynch II syndrome) due to germline inheritance of predisposing genetic abnormalities. However, most ovarian cancers are not thought to be due to inherited genetic alterations, but result from somatic mutations occurring in ovarian cells with an initially normal genome. These somatic mutations are thought to be secondary to environmental factors that act to increase the opportunity for spontaneous mutation in a number of critical genes. One of the genetic alterations that lead to cervical cancer is caused by an environmental factor, human papilloma virus (HPV), and in at least some endometrial cancers, unopposed oestrogen exposure or hyperinsulinism leads to carcinogenesis. High-risk subtypes of HPV are much less common in vulval cancer. This suggests that vulval and cervical carcinomas are not identical aetiologically, and that factors other than HPV are more important in vulval carcinogenesis. There is recent evidence that different genetic events occur between HPV-positive and -negative vulval cancers, with a greater number of molecular alterations in HPV-negative vulval cancers compared with HPV-positive tumours (Flowers et al 1999). HPV-positive vulval cancers are more frequently found in younger patients. Vulval cancers in older patients are more often HPV negative, and more frequently show allelic loss of the TP53 gene.
The histological features, and biological and clinical behaviour of fallopian tube cancers are similar to those of ovarian cancer. In addition, there is evidence that similar genetic alterations occur with the same frequency in fallopian tube and ovarian cancers of the same histological subtype. This suggests a common molecular pathogenesis between these cancer types (Levanon et al 2008).
To date, little is known about the molecular genetics of vaginal cancer.
Molecular Basis of Carcinogenesis
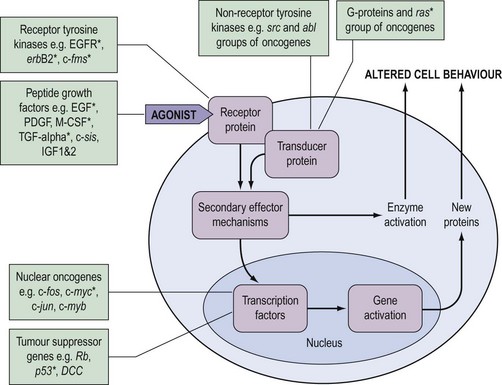
During the last decade, it has become clear that the genetic changes in cancer involve genes which control normal cell growth and proliferation. Cells communicate via a complex interacting set of signalling pathways, the principles of which, unlike the details, are well established (Figure 35.2). The best understood mechanism of communication between cells is via the release of molecules which interact as agonists with receptors either within the cell or, more frequently, in the cell membrane. This interaction results in a further signal within the cell which, via transducer proteins and secondary mechanisms, results in modified cell behaviour either by activating already synthesized target enzymes directly or by initiating transcription of genes and synthesis of their protein product. The genetic abnormalities observed in cancer are known to affect genes coding for all of the groups of molecules in Figure 35.2.
Oncogenes
The identification of oncogenes resulted from in-vitro and animal work with virus-induced cancers. As the viral genome is relatively small, it was possible to identify the specific genes (viral oncogenes) involved in the transforming activity of retroviruses, such as the Rous sarcoma virus. It was possible to identify homologous genes in normal cells, and a number of viral oncogenes were found to have normal cellular homologues known as ‘proto-oncogenes’. Other workers identified oncogenes by transfecting cell lines with DNA from human cancer cell lines, and found that some of the transforming oncogenes identified were mutated versions of the proto-oncogene identified by the retroviral approach. Subsequently, it was demonstrated that chromosome translocations such as the Philadelphia chromosome in chronic myeloid leukaemia involved known oncogenes (de Klein et al 1982).
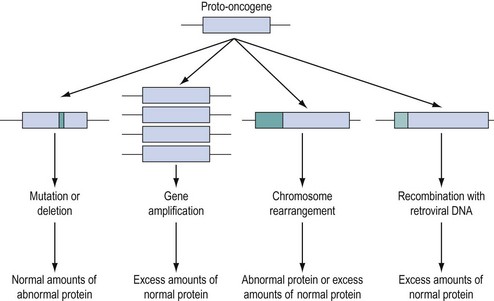
Approximately 100 proto-oncogenes have been identified, and many can be assigned to the groups of functions summarized in Figure 35.2. The mutation resulting in conversion of a proto-oncogene into an oncogene may occur in several different ways (Figure 35.3), and may result in an abnormal protein produced in a normal quantity, a normal protein produced in an excessive amount, or a normal protein produced inappropriately due to loss or alteration of control regions of the gene.
Tumour suppressor genes
The existence of tumour suppressor genes was suggested by cell fusion studies of transformed cells and non-transformed cells which resulted in a non-transformed hybrid cell. The subsequent identification of the first tumour suppressor gene followed from the study of retinoblastoma, a rare childhood cancer which may occur in a hereditary and sporadic form. On the basis of epidemiological and statistical analysis, Knudson suggested that development of the cancer required two events. He postulated that in the hereditary form, the first event was a germline mutation present in every cell of the individual, and that a second event in any of the many million retinoblasts could result in tumour formation. The rarity, later onset and unilateral nature of the sporadic form could be attributed to the need for two events in a single retinal cell in the absence of a germline mutation. When the retinoblastoma (Rb) gene was mapped to chromosome 13q and cloned, it was confirmed that in both familial and sporadic retinoblastoma, the two copies of the gene were inactivated in tumour cells, consistent with Knudson’s ‘two-hit’ hypothesis (Cavenee et al 1983).
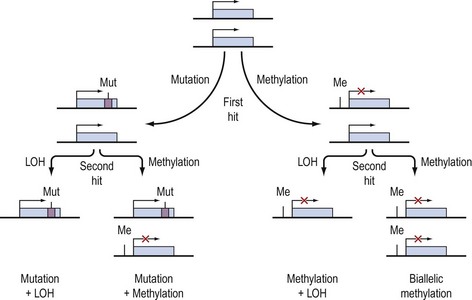
Knudson’s hypothesis that two hits are required for the full inactivation of a tumour suppressor gene has been shown to be fundamentally correct in almost all cases of human cancer. However, the fact that methylation of CpG islands located in the promoters of genes can cause transcriptional silencing, coupled with the observation that DNA methylation patterns are perturbed in cancer cells, has led to the suggestion that abnormal methylation of the promoters of tumour suppressor genes might be implicated in carcinogenesis, and can serve as a third pathway to inactivation of a tumour suppressor gene (Figure 35.4) (Jones and Laird 1999).
Epigenetics and DNA methylation in cancer
As outlined above, aberrant gene function and altered patterns of gene expression are key features of cancer. Growing evidence shows that acquired epigenetic abnormalities participate with genetic alterations to cause this dysregulation. The modern definition of epigenetics is modifications of the DNA or associated proteins, other than DNA sequence variation, that carry information content during cell division. The best understood example of epigenetic modification is DNA methylation, a covalent addition of a methyl group derived from S-adenosyl-L-methionine to the fifth carbon of the cytosine ring to form the fifth base, 5-methylcytosine (5meC) (Laird 2003, Jones and Baylin 2007). The reaction is catalysed by DNA methyltransferases together with accessory proteins. Among all eukaryotic species, methylation occurs predominantly in cytosines located 5′ of guanines, and known as ‘CpG dinucleotides’ (CpGs). In the mammalian genome, the distribution of CpGs is far from random. CpGs are greatly under-represented in the genome through evolutionary loss of 5meC through deamination to thymine. However, clusters of CpGs known as ‘CpG islands’ (CGIs) are present in 1–2% of the genome. Typically, they range in length from 200 to 5000 base pairs (bp). Most are unmethylated under normal circumstances, with the exception of those associated with imprinted genes, genes subjected to X-chromosome inactivation and transposable elements. In this last respect, DNA methylation is thought to repress faulty expression of endogenous transposons that may disrupt the genome. It is also involved in the parental-specific silencing of one allele of imprinted genes. In addition, approximately 70% of CGIs are associated with DNA sequences 200–2000 bp long found in the promoter, the first and second exons, and the first intron regions of all genes (5′ CGIs). This suggests that CGIs are critical in gene regulation. There is, for instance, usually an inverse relationship between the degree of methylation of a regulatory CGI and the extent of gene transcription (Figure 35.5) (Laird 2003, Jones and Baylin 2007).
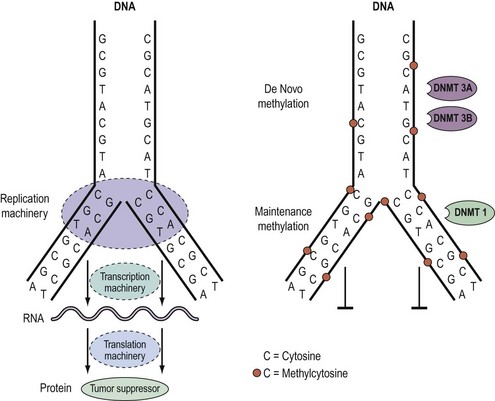
DNA methylation patterns are established during defined phases in embryonic development. With the exception of imprinted genes, gamete methylation patterns are erased by a genome-wide demethylation at around the eight-cell stage of blastocyst formation. During the implantation stage, methylation patterns are re-established via de-novo methylation. During adulthood, the degree and patterns of methylation are both tissue- and cell-type specific. Disruption of these preset patterns of DNA methylation in adult life has been linked to ageing and to disease. Furthermore, dysregulation of developmental programming by maternal factors or environmental mimics is thought to induce abnormal DNA methylation of specific genes and hence their faulty expression, leading to disease (Laird 2003, Jones and Baylin 2007).
Over the last decade, the importance of epigenetic gene regulation has become increasingly evident as studies have shown that each epithelial tumour only contains approximately 10–15 tumour suppressor genes silenced through mutations, but several hundred silenced through DNA methylation (Jones and Baylin 2007, Thomas et al 2007). Numerous genes involved in central pathways can be silenced by means of DNA methylation (Figure 35.3).
Germline and somatic genetic alterations
A common feature of tumours is that they accumulate somatic genetic changes during the development of the cancer. A proportion of cancers may also develop as a result of germline mutations in cancer predisposing genes. An important distinction must be made between germline and somatic mutations. Somatic mutations are found in the tumour cells but not the normal cells of an individual with cancer, and cannot therefore be passed on to descendants through the germline. Germline mutations, however, are present in all the cells of the individual and can initiate tumour development. They can be passed on to descendants of the individual, and can be identified in DNA obtained from all diploid cell populations (e.g. peripheral blood samples) and not just the tissues that are susceptible to disease. It is worth pointing out that the same genes mutated in the germline are also frequently mutated in the somatic cells of a cancer, which suggests a functional significance for specific genes in cancer development. For example, mutations of the p53 gene are one of the most common genetic changes found in human cancer. In most cancers, p53 mutations occur as somatic events, but germline p53 mutations are often found in patients from families with the rare Li-Fraumeni syndrome, which is characterized by sarcomas, breast cancer and other malignancies occurring at a young age. Another example is the colorectal cancer predisposition syndrome, familial adenomatous polyposis (FAP), caused by germline mutations in the APC gene (Kinzler et al 1991). The FAP syndrome is responsible for approximately 1% of all colorectal cancers, but somatic mutations of APC have been detected in over 80% of sporadic colorectal cancers and appear to be one of the earliest events in colorectal carcinogenesis.
Most of the familial cancer syndromes identified to date are rare, single gene disorders and have an autosomal-dominant pattern of inheritance. Individuals inheriting the germline abnormality are generally at high risk of developing malignancy because the penetrance of most of these genes is of the order of 80%. Another class of cancer predisposition genes that confer more modest penetrance has been identified recently. Although these genes are associated with much lower penetrance, they are also much more common in the population than the rare high-risk genes, and so are probably responsible for a greater proportion of cancer overall (reviewed in Easton and Eeles 2008). However, the identification of individuals in the population that are likely to carry these low penetrance genes is not easy because they are not often associated with a significant family history of disease.
Genetics of Familial Gynaecological Cancer
Although there are anecdotal reports of more than one case of vulval cancer occurring in close relatives, there is no good evidence of inherited predisposition to this disease. For cervical cancer, the absence of any obvious clustering of multiple cases within families suggests that no highly penetrant susceptibility loci for the disease await identification. However, population-based epidemiological studies suggest that shared genetic factors do play a role in the development of cervical cancer, and possibly account for approximately 27% of cases. This perhaps suggests the presence of several common genetic variants that confer low penetrance disease risk. Ovarian and endometrial cancers occur as part of well-defined cancer family syndromes with an autosomal-dominant inheritance pattern. Pedigree studies of cancer within families have revealed three main syndromes associated with ovarian cancer. The most common hereditary form of ovarian cancer occurs in association with breast cancer. A large number of hereditary breast/ovarian cancer families have been described in which there is a high frequency of both cancers and an association with an early age of onset. Hereditary ovarian cancer may also occur as a site-specific disease, although genetic studies suggest that a large proportion of site-specific ovarian and breast/ovarian cancer families are part of the same disease spectrum. Nevertheless, there remains evidence that there is a rare genetic component that confers high penetrance susceptibility specifically to ovarian cancer (reviewed in Ramus and Gayther 2009). Less frequently, ovarian cancer occurs in hereditary non-polyposis colorectal cancer (HNPCC) families, and germline genetic mutations in highly penetrant genes that cause HNPCC have been found in ovarian cancer cases from these families. Women from HNPCC families most frequently develop colorectal or endometrial cancer, and this is the syndrome most commonly associated with genetic predisposition to endometrial cancer. Some families have recently been described with an apparently high risk of site-specific endometrial cancer. It is important to recognize that when all of these familial syndromes are combined, they are probably responsible for less than 5% of ovarian cancer cases and a smaller proportion of endometrial cancer cases.
The BRCA1 and BRCA2 genes
The identification and detailed pedigree-based descriptions of families at high risk of breast and ovarian cancer, together with the development of polymorphic DNA markers, made it possible to perform detailed linkage analysis with the aim of locating genes involved in familial cancer. A major step forward was the report of linkage of early-onset breast cancer to the polymorphic marker CMM86 located on chromosome 17q (Hall et al 1990). Subsequently, linkage to 17q in 214 families was confirmed by a consortium of 13 research groups from Europe and the USA (Easton et al 1993). Almost all breast/ovarian cancer families and 40% of families with breast cancer alone were found to have a linkage with the BRCA1 (Breast Cancer 1) locus on chromosome 17q. Subsequently, the consortium localized the gene to a 2cM region on 17q, and in 1994, a group in Utah described a large gene on 17q within which they had identified mutations in five affected families (Miki et al 1994). Subsequent work by other research groups has confirmed that this gene is the BRCA1 gene, and over 12,000 carriers of a BRCA1 mutation have now been described in breast/ovarian cancer families and cancer cases unselected for a family history [described on the Breast Cancer Information Core (BIC) database: http://research.nhgri.nih.gov/bic/].
BRCA1 is a large gene with 22 exons encoding a protein of 1863 amino acids. There are no specific hotspots for mutation in the gene, and only a small proportion of mutations are recurrent. Overall, the penetrance of a BRCA1 mutation for ovarian cancer and breast cancer appears to be 40–50% and 80%, respectively Ford et al 1994, 1998). However, there is evidence that penetrance may be modified by both environmental factors and other genetic influences. Furthermore, the location of a mutation within the BRCA1 gene may influence both the overall risk of cancer and the type of cancer. Gayther et al (1995) reported a correlation between mutations toward the 3′ end of the gene and a greater risk of ovarian cancer than breast cancer. Well-defined founder mutations have been described in BRCA1. Mutations at 185delAG and 5382insC have been documented in approximately 1% of Ashkenazi Jewish cancers, and up to 40% of Ashkenazi Jewish women with ovarian cancer or early-onset breast cancer. Identification of BRCA2 closely followed the cloning of BRCA1. Wooster et al (1995) identified the gene following studies involving high-risk families in which the disease pattern was not linked to BRCA1.
BRCA2, located on chromosome 13q, is also a large gene with 26 coding exons encoding a protein with 3418 amino acids. More than 11,000 mutation carriers have been described to date (the BIC database). As for BRCA1, there are no hotspots for mutation, although there are founder mutations in some ethnic groups such as Ashkenazi Jews (Levy-Lahad et al 1997, Thorlacius et al 1997). Although the penetrance for breast cancer is approximately 80%, the penetrance for ovarian cancer seems to be lower than that for BRCA1 mutations at approximately 25% (Ford et al 1998). There is a suggestion that mutations in exon 11 confer a higher risk of ovarian cancer than mutations in other areas (Gayther et al 1997). This has been confirmed in a more recent meta-analysis (reviewed in Ramus and Gayther 2009). Overall, BRCA1 and BRCA2 are believed to account for over 95% of hereditary cases of ovarian cancer and over 80% of hereditary cases of breast cancers.
The functions of BRCA1 and BRCA2 are still unclear. Most of the mutations of BRCA1 are small insertions or deletions, which result in loss of protein synthesis or production of a truncated protein. This observation supports the concept that BRCA1 functions as a tumour suppressor gene. In this ‘two-hit’ model, although one copy of the gene is inherited as an inactive mutant form, the development of cancer requires somatic mutation of the other wild-type (‘normal’) gene copy. Analysis of tumours from familial breast/ovarian cancer families has demonstrated that loss of heterozygosity (LOH) in these tumours involves the wild-type gene, thus providing further evidence that the BRCA1 gene is a tumour suppressor (Smith et al 1992). Both BRCA1 and BRCA2 are expressed in the largest amounts in the testis and thymus, and at lower levels in the breast and ovary. Although the genes have limited sequence homology, they do have similarities including being A-T rich, having a large exon 11 with the start of translation at codon 2. The presence of a zinc finger motif in BRCA1 has suggested a role as a transcription factor, whilst BRCA2 has homology with known transcription factors. However, the most compelling functional evidence indicates that BRCA1 and BRCA2 both play a major role in homologous and non-homologous DNA double-strand break (DSB) repair through their interaction with the RAD51 DSB protein. It is still not known why abrogation of such a ubiquitous cellular function should predispose individuals specifically to breast and ovarian cancer.
HNPCC-associated gynaecological cancer and DNA repair genes
HNPCC is associated with autosomal-dominant inheritance of colorectal cancer in those who do not have the multiple adenomas which occur in FAP. The identification of the genetic basis of Lynch II syndrome is a notable example of the power of molecular technology. HNPCC was classified by Henry Lynch into site-specific hereditary colon cancer (Lynch I syndrome), and families with a predisposition to non-polyposis colorectal cancer in association with other cancers including stomach, small bowel, ureter, renal pelvis and brain, as well as ovarian and endometrial cancer (Lynch II syndrome) (Lynch 1985). The identification of the genes responsible for HNPCC was a result of several developments which showed that HNPCC is associated with hereditary defects in one of five DNA mismatch repair genes [mutS homolog 2 (MSH2), mutL homolog 1 (MLH1), postmeiotic segregation increased 1 (PMS1), postmeiotic segregation increased 2 (PMS2) and mutS homolog 6/G/T mismatch-binding protein (MSH6/GTBP)]. Hundreds of mutations have been identified in these five genes in HNPCC families, making mutation testing a difficult challenge in these families (Lynch and de la Chapelle 1999). Available evidence suggests that these genes function as tumour suppressor genes in a manner consistent with Knudson’s ‘two-hit’ model. The inherited germline mutation inactivates one copy of the mismatch repair gene, but inactivation of the remaining wild-type copy by a second somatic mutation is required prior to tumour formation. The secondary, acquired mutations which result from loss of DNA repair gene function include defects of APC, K-ras, DPC4 and p53 (Huang et al 1996, Kinzler and Vogelstein 1996). The penetrance for colorectal cancer in mutation carriers is in excess of 80% in men and 30% in women, and HNPCC accounts for approximately 5% of colorectal cancers (Dunlop et al 1997, Aarnio et al 1999, Salovaara et al 2000). Endometrial cancer is the second most common cancer in HNPCC families, with female carriers having a 42% risk by 70 years of age (Dunlop et al 1997). The penetrance for ovarian cancer is less, with a reported lifetime risk of 9% (Marra and Roland 1995).
Management of Familial Gynaecological Cancer
Risk assessment
Prevention of familial cancer
Use of the oral contraceptive pill may be suggested for HNPCC and breast/ovarian cancer family members on the basis of case–control studies in the general population which indicate a protective effect against ovarian and endometrial cancer. While one study has suggested a protective effect of the oral contraceptive pill in BRCA1 and BRCA2 carriers, this was not confirmed in a second study and the situation remains unclear (Modan et al 2001). Furthermore, there is concern in breast/ovarian cancer families that a reduction in risk of ovarian cancer may be offset by an increased risk of breast cancer.
Prophylactic salpingo-oophorectomy and hysterectomy as a primary procedure is justifiable in women from breast/ovarian cancer and HNPCC families after completion of their family and after thorough counselling. There is convincing evidence that salpingo-oophorectomy is an effective method of prevention of cancer of the ovary, fallopian tube and breast. A recent meta-analysis (Rebbeck et al 2009) of all reports of preventative surgery in BRCA1 and BRCA2 mutation carriers from 1999 to 2007 revealed a reduction in risk of breast cancer (hazard ratio 0.49) and ovarian/fallopian tube cancer (hazard ratio 0.21). Women undergoing prophylactic surgery should be counselled that the procedure will prevent ovarian and tubal cancer, but that they may still be at risk of primary peritoneal cancer.
It should also be noted that cases of intra-abdominal carcinomatosis following oophorectomy have been reported in which subsequent review of the oophorectomy specimen revealed a small focus of ovarian cancer (Chen et al 1985
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


