A large number of children with epilepsy will develop SE at least once. A community-based cohort of 613 children with epilepsy in Connecticut reported that 7.3% had at least 1 episode of unprovoked SE. Further, in 16 of the 45 children, SE was their initial seizure type (22). A Dutch cohort reported similar numbers, with 9.5% of children with epilepsy having at least 1 episode of unprovoked SE (23). However a Finnish population based study reports a much higher incidence of 27% (24). The greatest risk for prolonged seizures is during the first 2 years after diagnosis of epilepsy (24). Other risk factors for SE in children with preexisting epilepsy include focal seizures and/or focal electroencephalography (EEG) abnormalities (odds ratio [OR], 2.2–6.51), recent intracranial procedures (OR, 22.1), and presentation with SE as their first seizure (22,25). Children with some specific epilepsy syndromes, including Dravet syndrome (severe myoclonic epilepsy of infancy) and Panayiotopoulos syndrome (benign childhood autonomic epilepsy), are at higher risk of prolonged seizures (Figure 8.1) (15). Genetics may contribute to risk of SE as well; monozygotic twins have a higher concordance rate for SE than dizygotic twins (26).
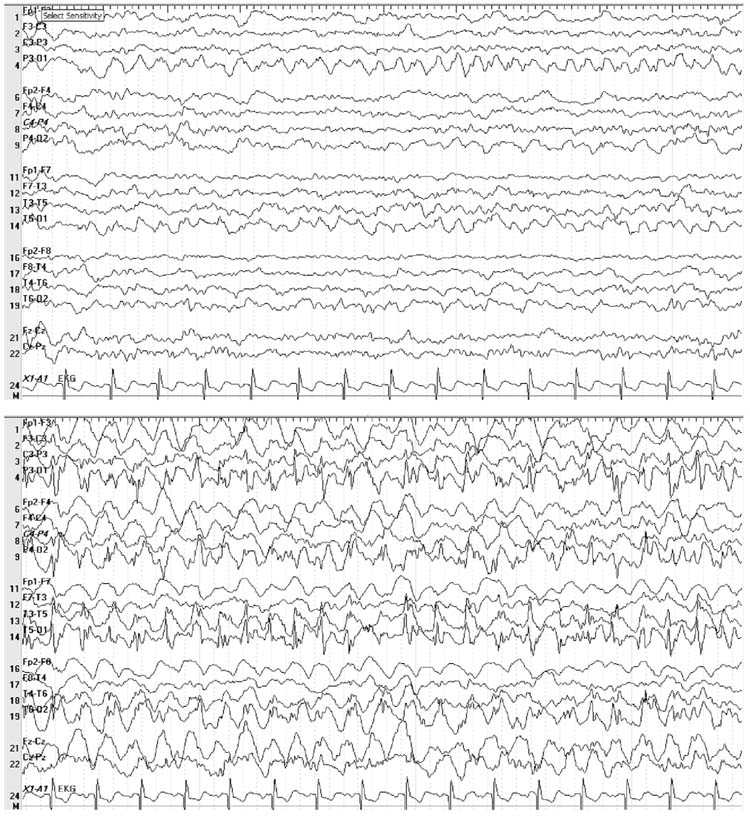
FIGURE 8.1 Five-year-old boy with a history of febrile seizures admitted with refractory status epilepticus with clinical seizures characterized by right sided jerking that required a midazolam drip for control. Electroencephalogram (EEG) showed occipital lobe seizures and stereotyped occipital discharges consistent with a diagnosis of benign occipital epilepsy of childhood. This image shows a left occipital lobe seizure (displayed at a sensitivity of 15 μV) that spreads to the right occipital lobe as the seizure progresses.
Many children that present with SE do not have a history of seizures or epilepsy. Singh et al (13) reported that 10% of children present with SE as their first seizure. In the ICU, the percentage of children with SE as their first seizure is even higher; one study found that 71% of children admitted to the pediatric ICU (PICU) with a diagnosis of SE do not have a prior history of seizures (20). The discrepancy in these numbers may be due to the bias towards more serious acute symptomatic etiologies in critical care populations.
In addition, children with SE often require care in the PICU (20,27–29). A retrospective review of admissions to a tertiary care hospital PICU in Canada revealed that 1.6% of all admissions between 1976 and 1986 were for SE (29). The absolute number of admissions varies between tertiary care centers due to location and size of the ICU. Over a 5 year period in three different institutions, the absolute number of reported admissions for SE ranged from 25 to 218 (20,27,28). Admission to the PICU for any type of seizure is even higher; a total of 554 admissions to the PICU were reported over a 5 year period at one tertiary care center (20). Of note, these numbers do not include the number of children who developed SE or seizures after admission to the hospital.
The true incidence of seizures and SE in the PICU is even higher when nonconvulsive seizures are included. Over the last decade, the use of continuous EEG has led to an increased awareness of the prevalence of nonconvulsive seizures and nonconvulsive SE in critically ill children. Some children have preceding clinical seizures (Figure 8.2) but others may present only with nonspecific changes in mental status or coma. In a retrospective study of 236 patients admitted to the ICU with coma (11% were children aged 1 month through 15 years), 8% of patients had nonconvulsive SE (30). In another large retrospective study at a tertiary care hospital, 16.3% of children monitored with video EEG over 3 years had nonconvulsive seizures; none of these children presented first with convulsive seizures during that admission and 43% had no preexisting neurologic conditions (31). Similarly, 2 studies concluded that approximately 45% of children monitored by continuous EEG have nonconvulsive seizures and 69% to 75% of them only have electrographic seizures, and thus would not be detected without EEG monitoring. Common etiologies for isolated electrographic seizures include acute infarcts, cerebral edema, and diffuse hypoxic-ischemic encephalopathy (32,33). Even these numbers may underestimate the incidence of nonconvulsive seizures because some electrographic seizures are detectable only on intracortical EEG (34).
FIGURE 8.2 Seven-week-old boy with cardiorespiratory failure due to pertussis who was placed on extracorporeal membrane oxygenation. Patient initially had clinical seizures with right hand clonic movements. After convulsive seizures abated, continuous EEG (sensitivity 5 μV) revealed nonconvulsive seizures from the right frontal lobe that required a midazolam drip for cessation. MRI brain done 1 month later showed diffuse atrophy, ex vacuo hydrocephalus, and focal encephalomalacia on axial FLAIR (fluid-attenuated inversion recovery) images (A) and multifocal gradient susceptibility consistent with prior hemorrhage, most notable in the right frontal lobe (B).
■ CRITICALLY ILL CHILDREN WITH INCREASED RISK FOR SEIZURES
Hypoxic-Ischemic Encephalopathy
Hypoxic injury is a common cause for SE in adults; 15% to 44% of adults with cardiac arrest will have seizures or postanoxic myoclonus (35). Anoxia is a less common reason for seizures in children, likely due to the lower incidence of cardiac arrest in the PICU than in adult ICU (6). Nonetheless, children with global hypoxia-anoxia (e.g., cardiopulmonary arrest) are at increased risk for seizures. EEG monitoring of 19 children during hypothermia after cardiac arrest revealed that 47% of the patients developed seizures, approximately two-thirds of which were nonconvulsive (36).
Extracorporeal Membrane Oxygenation
Children placed on extracorporeal membrane oxygenation (ECMO) are an important group of patients in the PICU that are at high risk for seizures. Complications of ECMO include intracranial hemorrhage, infarction, hypoperfusion, microemboli, and electrolyte abnormalities which all may be symptomatic etiologies for seizures. Furthermore, children who require ECMO usually have a diagnosis that increases the risk of seizures such as cardiac failure, respiratory failure, or sepsis. Approximately 40% of children who survive ECMO as a neonate have a history of acute symptomatic seizures (37). One study reported that 11.5% of pediatric patients older than 1 month of age placed on ECMO developed seizures during the acute period (38). However, the incidence of seizures was higher in children with sepsis as well, so it is difficult to discern the effect of ECMO versus infection as risk factors for seizures (38).
Traumatic Brain Injury
Children are twice as likely as adults to experience early posttraumatic seizures, which are defined as seizures occurring less than 7 days after injury (39). The risk of seizures rises with increasing severity of injury, from 1% to 2% in mild traumatic brain injury to 20% to 39% in severe injuries (39–43). Children under 2 years of age are at greatest risk for developing seizures (OR, 2.96 for children < 2 years compared to children > 12 years of age) (43). Early recognition and treatment of seizures is important to minimize cerebral metabolic demand, prevent hyperthermia, and prevent elevation of intracranial pressure. The use of prophylactic antiepileptics (AEDs) to prevent early posttraumatic seizures in pediatric patients “may be considered” in severe traumatic brain injury according to a consensus guideline; however prophylactic AEDs are not recommended to prevent late posttraumatic seizures (after 7 days from injury) as there is no evidence that this tactic is effective (44). During early resuscitation in the PICU, controlling seizures can improve management and stability, but beyond early stabilization, this approach is likely unnecessary (45).
Children with a history of nonaccidental (inflicted) trauma are at particularly high risk for posttraumatic seizures, possibly due to the multiple mechanisms of possible injury (shear injury, diffuse axonal injury, ischemic infarct from vascular injury, hemorrhage, contusions, anoxia). In a study of 14 children with varying degrees of intracranial injury, 79% had posttraumatic seizures within the first 72 hours of admission. Seizures in 8 of the children were considered “difficult to control” (46). A large French study also reported that 73% of children with shaken baby syndrome presented with or developed seizures during the acute period (47).
Infection
One of the most common neurologic complications of CNS infections is acute symptomatic seizures. In addition to direct invasion by the pathogen and increased inflammation, CNS infection increases the risk of seizures by causing intracranial abnormalities (eg, hydrocephalus, abscess, ischemia, hemorrhage, sinus venous thrombosis), electrolyte abnormalities (eg, hyponatremia from syndrome of inappropriate antidiuretic hormone hypersecretion, cerebral salt wasting), and/or fever. Approximately a quarter of patients with any type of CNS infection (meningitis, encephalitis, brain abscess, empyema) develop provoked seizures (48,49). Thirty-one percent to 50% of infants and children have seizures during acute bacterial meningitis (Figure 8.3) and one study reported that 13% of children in the PICU with meningitis developed SE (29). Acute seizures in bacterial meningitis are predictive of poor outcome and as is the development of late seizures (50,51). One contributing factor to these findings is that children with acute seizures are more likely to have structural lesions than those without seizures (50,51).
FIGURE 8.3 Twenty-month-old boy with meningitis who developed refractory status epilepticus. Patient initially had clinical seizures with nystagmus, upper extremity jerking which evolved into subtle clinical seizures with nystagmus alone. After clinical seizures stopped on a midazolam drip, continuous EEG (sensitivity 15 μV) shows right occipital lobe spikes and slow wave discharges consistent with an electrographic seizure; (2) MRI of the brain with and without contrast revealed on axial FLAIR images, diffuse patchy areas of increased signal in the subcortical and periventricular white matter, internal and external capsules, and thalami (A) and on axial postcontrast T1 images, diffuse leptomeningeal enhancement (B).
Seizures and SE also frequently occur with viral encephalitis. In one study, 40% of children in the PICU with viral encephalitis developed SE (29). Most of the research on seizures in the setting of viral encephalitis focuses on herpes simplex virus (HSV) encephalitis, which has a predilection for the temporal lobe. Most studies estimate that 40% to 60% of patients with HSV will have seizures acutely (52,53). However, one case series from India reported that as many as 89% of patients will have seizures (54). The predictive value of provoked seizures in children with viral encephalitis is controversial. Some studies conclude that multiple acute symptomatic seizures and SE are poor prognostic factors in children with viral encephalitis whereas others researchers report that there is no difference in outcome between patients with and without seizures (55,56).
Influenza infections are frequently seen in the PICU, and a small percentage of children with influenza, with or without encephalopathy, develop seizures. One study reported that 6.5% of children admitted with influenza over a 4 year period developed seizures; approximately 50% of them were deemed febrile seizures, 14% were acute symptomatic, and 37% occurred in children with preexisting neurologic problems (remote symptomatic with acute provocation) (57). A similar incidence (6%) of seizures was reported in children admitted to a Chinese hospital in 2009 with H1N1; children in the ICU are more likely to experience seizures, presumably because these children are the sickest (58). However, the overall incidence of seizures with influenza infection is lower than reported in these studies since most children do not require admission for influenza infection; a Morbidity and Mortality Weekly Report from 2009 reported that only 2.7% of children in Dallas with influenza had acute symptomatic seizures, 50% of which were due to influenza encephalopathy (59).
The risk of seizures is not confined to the period of acute infection, and may extend to the postinfectious period. Seventeen percent to 47% of children with acute disseminated encephalomyelitis have seizures at some point in their disease course and many present with seizures. One study has documented prolonged focal motor seizures in 70% of the younger patients, with 82% of these patients going on to develop SE (60,61).
Cerebrovascular Disease
Stroke is less prevalent in children than in adults; however, children are more likely to present with seizures or develop early seizures after cerebral ischemia (Figure 8.4). A population based study in the Greater Cincinnati area reported that 3.1% of adults experienced acute seizures with ischemic stroke; in contrast, a population based study of children (excluding neonates) in the same region reported an incidence of acute seizures in 58% of children (62,63). The incidence of seizures is higher with hemorrhagic than ischemic stroke.
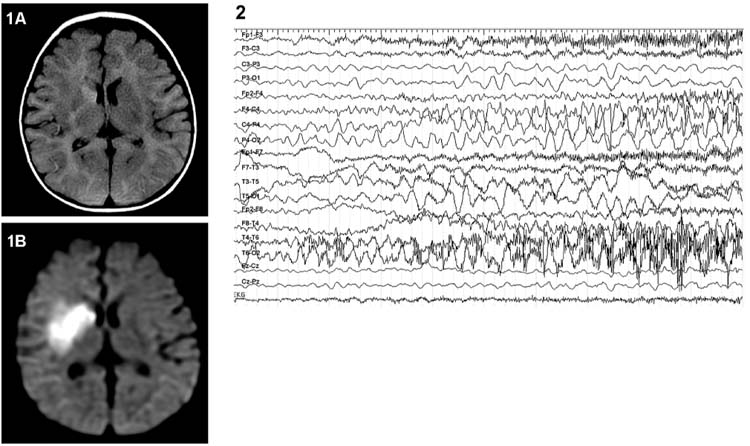
FIGURE 8.4 Six-month-old girl with a right middle cerebral artery stroke with periods of decreased responsiveness. MRI imaging showed (1), Increased FLAIR signal in the right basal ganglia (A) and diffusion restriction in a right middle cerebral artery distribution (B). (2), Continuous EEG monitoring (sensitivity of 20 μV) found that these episodes correlated with electrographic seizures.
Children with sickle cell disease are a high risk group for cerebrovascular disease including ischemic stroke, hemorrhage stroke, transient ischemic attacks, and cerebral sinus venous thrombosis. They are approximately 10 times more likely than the general population to have seizures (12%–14% of patients) (64–66). Children with seizures are more likely to have cerebral hypoperfusion and vasculopathy. Seizures can be secondary to remote symptomatic epilepsy related to prior cerebral injury but in up to one-third of sickle cell patients, seizures are the presenting symptom of acute stroke (65).
Children with cerebral venous thrombosis also are at high risk for seizures. The seizures may be the heralding sign of a cerebral venous thrombosis in approximately 30% to 50% of adults (67–69). In several multicenter studies, another 7% to 10% of patients had early seizures within 2 weeks of diagnosis (67–69). Studies report a higher incidence of seizures as a presenting symptom in infants than children with cerebral venous thrombosis (50%–60%) (70–73). Since a large percentage of critically ill children have risk factors for cerebral venous thrombosis (eg, dehydration, infection), clinicians should consider cerebral venous thrombosis as a possible etiology for new onset seizures in the PICU.
Congenital Heart Defects
Children with congenital heart defects often have multiple risk factors for seizure including chronic hypoxia with risk of acute hypoxia during bypass surgery or periods of cardiac decompensation, hemorrhage, prematurity, thromboemboli, and metabolic disturbances. They are most vulnerable during the postoperative period. A study in San Francisco found that 13% of 114 infants had EEG seizures in the postoperative period after repair of a congenital heart defect; 22% of infants with hypoplastic left heart syndrome had postoperative seizures (74). A similar incidence is reported in a Philadelphia cohort and the Boston Circulatory Arrest study (75,76). Nonconvulsive seizures also occur frequently in this patient population (31).
Hypertensive Encephalopathy
Hypertension is a common problem in the critical care unit, and is often secondary to medications (steroids, chemotherapy/immunosuppressant medications), renal disease, or other primary medical problems. Children with hypertensive encephalopathy or posterior reversible encephalopathy syndrome often present with seizures along with encephalopathy, visual changes, headache, or other neurologic dysfunction (Figure 8.5). Seizures may be single, multiple, focal, or generalized (77,78).
FIGURE 8.5 Three-year-old girl status post stem cell transplant for clear cell carcinoma and central nervous system metastases who presented hypertension after cyclosporine, altered mental status, and visual problems. Imaging findings were compatible with a diagnosis of posterior reversible encephalopathy syndrome. (1), MRI of the brain: T2 sequence shows areas of increased signal in the bilateral parietal occipital lobes, predominantly on the right side (A); diffusion weight imaging also shows diffusion restriction in a similar distribution (B). (2), Continuous EEG monitoring (10–20 international system convention) shows right occipital electrographic seizures without clinical correlate (displayed at sensitivity of 15 μV).
■ TREATMENT
Evaluation of the patient’s vital signs and an assessment of airway, breathing, and circulation (with appropriate interventions) should occur at the same time as AED treatment is initiated. Oxygenation should be maintained with supplemental oxygen when appropriate. There is no consensus as to a treatment approach for SE with respect to either medication selection or dosing, and therapy is highly variable between institutions and practitioners. A guiding general principle in the medical treatment of SE with AEDs is to rapidly administer appropriate medications at appropriate doses. Common errors include medication underdosing, excessive intervals between medications, and inappropriate medication choices and routes of administration. AEDs may impact the airway, breathing, and circulation, so close monitoring is essential. Our management pathway is provided in Figures 8.6A and 8.6B.
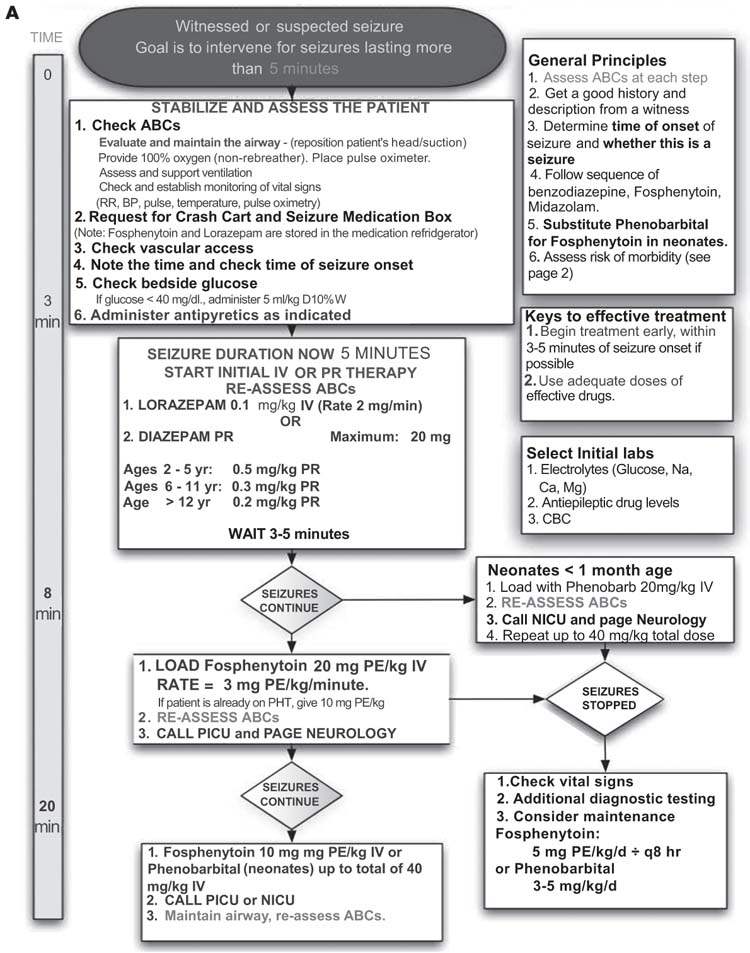
FIGURE 8.6 (A) Inpatient Guidelines for management and evaluation of status epilipticus. (Continued.)
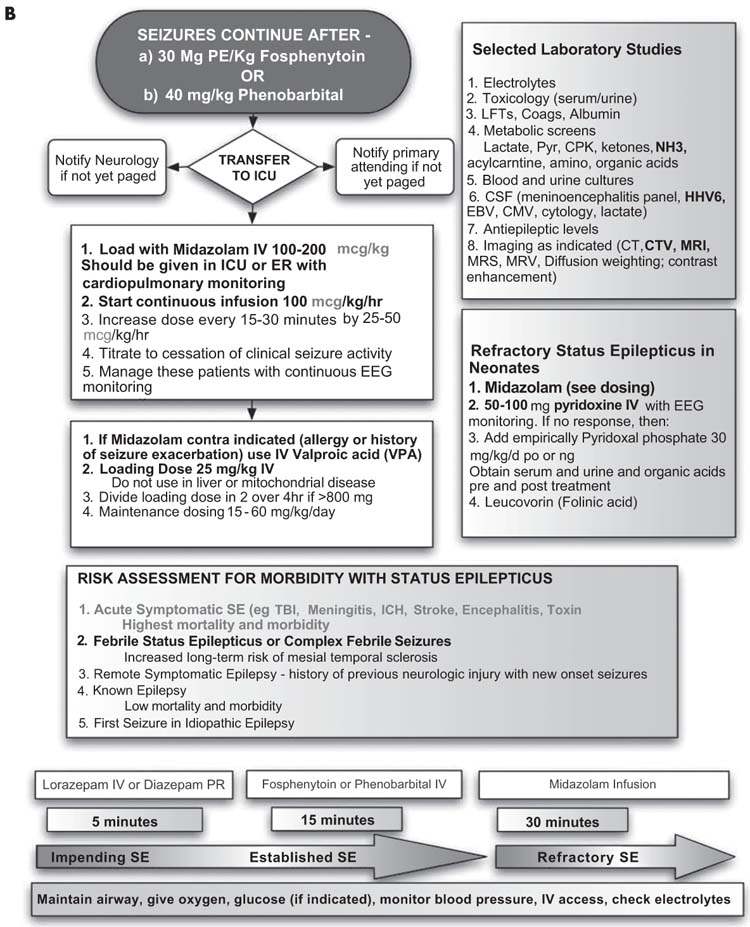
FIGURE 8.6 (B) refractory status epilipticus.
Benzodiazepines including diazepam, midazolam, and lorazepam are the most commonly used first-line AEDs for SE. Intravenous or rectal routes of administration are usually preferred for these drugs given their more rapid onset of action. Dosing recommendations are highly variable as are the number of repeated doses, although most will give 2 or 3 total doses of a benzodiazepine. Frequent dosing of small or inadequate individual doses should be avoided as this practice only prolongs the time to achieving a therapeutic level of the AED. Rather, higher individual doses should be used either once or twice to allow for the more timely institution of a second AED if needed.
Fosphenytoin is often considered the second AED for SE treatment. This prodrug of phenytoin converts to phenytoin within 15 minutes of infusion. Care in dosing is important as it is dosed in phenytoin equivalents (PE) rather than milligrams. A standard initial loading dose for SE is 20 PE/kg, although some use lower doses. Advantages of fosphenytoin over phenytoin include a lower risk of cardiac dysfunction with acute infusion, lower rates of phlebitis, and less severe tissue necrosis if the medication extravasates from the vein during infusion. Although it can be infused at a significantly faster rate than phenytoin, fosphenytoin should not be infused at a rate greater than 3 PE/kg/minute (or, over 7 minutes for a 20 PE unit/kg load). A loading dose of 20 PE units/kg often provides a serum phenytoin level of around 20 μg/mL. A second dose of 10 PE/kg (to a total of 30 PE/kg) is often given if the first loading dose fails to terminate SE. The rapidity of the administration of the AED is critical and delays should be strictly avoided. At our institution, we aim to administer fosphenytoin within 20 minutes of treatment initiation.
Phenobarbital is commonly chosen as the next AED for refractory SE given its long history of use, good efficacy, and the practitioner comfort level with it as a familiar medication. Phenobarbital is given via intravenous infusion at an initial dose of 20 mg/kg. While it is often highly effective, potential adverse effects include substantial respiratory suppression, especially after benzodiazepines have been given, often requiring ventilatory support. Phenobarbital’s half-life of 24 to 72 hours also makes the ongoing assessment of clinical neurologic function difficult for a prolonged period. For these reasons many clinicians are now avoiding its use in refractory SE, opting for some of the treatments discussed below. In the infant, many sites use phenobarbital in place of fosphenytoin as a second-line medication based primarily on its long history of use and not any formal study.
After appropriate doses of benzodiazepines and fosphenytoin and sometimes phenobarbital have been given, the selection of subsequent AEDs is even more varied. At this point, having persisting SE despite administration of adequate doses of two AEDs, the patient has met the criteria for refractory SE and a neurologist should be consulted if available. Commonly used treatments for refractory SE include valproic acid (Depacon), levetiracetam (Keppra), intravenous anesthetic agents (propofol), potent inhalational anesthetics (isoflurane), and continuous infusions of barbiturates (pentobarbital) or benzodiazepines (midazolam).
Midazolam continuous infusions are usually started at 50 to 100 μg/kg/h and titrated upwards to a range of 600 to 1,200 μg/kg/h. To avoid tachyphylaxis, the infusion rate increases should be aggressive, reaching a maximal dose a few hours after initiation. One commonly used protocol increases the rate by increments of 50 to 100 μg/kg/h every 15 to 20 minutes. Continuous EEG should be instituted if available when continuous infusions of AEDs are used to ensure that nonconvulsive seizures do not persist after convulsive seizures terminate.
Like midazolam, pentobarbital can also be administered as a continuous infusion for refractory SE. It is usually initiated with a loading dose of 6 to 8 mg/kg followed by a continuing infusion at a rate of 0.5 to 1 mg/kg/h and then increased as needed to 3 to 5 mg/kg/h. Pentobarbital, like phenobarbital, is extremely sedating and has a higher associated rate of hypotension than midazolam, often requiring pressor support.
Intravenous valproic acid and levetiracetam have some evidence for their efficacy in SE and many centers are investigating their use in SE treatment. There are some theoretical advantages to these medications. Valproic acid and levetiracetam are considered broad spectrum AEDs and should not exacerbate primary generalized epilepsy as can phenytoin or fosphenytoin. They also are considered less sedating than the benzodiazepines, phenobarbital, or phenytoin, thereby allowing for more precise ongoing neurologic examinations. Valproic acid is most commonly dosed intravenously between 20 and 60 mg/kg (79). A second dose may be given. Levetiracetam is also administered intravenously and doses range from 20 mg/kg to 60 mg/kg or higher (80,81).
The use of anesthetic agents in the treatment of refractory SE is rapidly gaining popularity in adult neurology but has yet to gain as widespread acceptance in the pediatric population (82,83). The most commonly used anesthetic is propofol. This medication has the advantage of a short half-life, allowing reexaminations. Propofol may cause “propofol syndrome” manifest by acidosis, rhabdomyolysis, and cardiac/renal failure. This is more frequent in children than adults, and this risk limits the duration propofol can be administered. Thiopental has also been utilized for refractory SE with good efficacy. As escalating doses of drugs are used to treat SE, often, protective airway reflexes are lost, and intubation and mechanical ventilation become necessary. For SE not responsive to the above AEDs, potent inhalational anesthetics such as isoflurane have been utilized (84,85).
■ PRIMARY SYSTEMIC COMPLICATIONS FROM SE
Seizures and SE cause local neuronal inflammation/injury, calcium related injury, cytotoxic injury, alterations in glutaminergic and γ-aminobutyric acid receptors, and other changes in the neuronal cell environment. These changes in the neuronal network, the activation of the autonomic nervous system, changes in metabolic state, and the strain of repetitive convulsions can lead to systemic complications (86–92).
The continuous contraction and relaxation of muscles with convulsive SE can lead to muscle breakdown, fever, rhabdomyolysis, and subsequent renal failure (93–97). Fever increases metabolic demand of the brain and augments excitotoxicity and neuroinflammation, worsening ongoing brain injury in acute symptomatic etiologies. Convulsions also increase metabolic demand in the muscle; eventually muscle changes from aerobic to anaerobic respiration, with the production of lactic acid and acidosis (98). Thus, maintenance of adequate hydration is extremely important. Ultimately, muscle relaxants and active cooling may be required. Seizures also alter autonomic and cardiac function. Some patients develop ictal tachycardia, ictal bradycardia, and postictal changes in heart rate variability, while some AEDs induce prolonged corrected QT interval (97,99,100).
Blood pressure usually rises in the acute stage of SE but falls with more prolonged convulsive seizure activity (usually after 30–40 minutes in animal models) (101, 102). Patients with SE also are at increased risk for cardiac failure and arrhythmias. Some patients may display gradual compromise of cardiovascular function with decreased blood pressure and/or heart rate prior to death but acute cardiac decompensation occurs in other patients (103). Cardiac myofilament damage, myocardiac stunning, and myocardiac contraction bands have been described in animal models of SE as well as autopsy of patients who died from SE (104–106). Cardiac injury is suspected to be secondary to unopposed sympathetic activation in SE.
Independent of medication side effects, seizures can affect respiratory status. Ictal hypoxia occurs in 40% or greater of children with generalized seizures (107–109). Neurogenic pulmonary edema is a less common complication (110,111).
Brain metabolism is affected by SE. Initially blood glucose rises with release of catecholamines but glucose normalizes or decreases after 30 to 40 minutes (101,112). Cerebrovascular reserve and blood flow may increase initially but these changes cannot be maintained over time (112–114). With prolonged seizures, cerebral energy demands remain high but stores of oxygen and glucose become depleted; this inequity contributes to cerebral injury in animal models (102,112). Cerebral edema can develop, likely due to the ongoing CNS inflammation but also from an increase in vascularity and compromise of the blood-brain barrier (113,115–117).
■ COMPLICATIONS SECONDARY TO SE TREATMENT
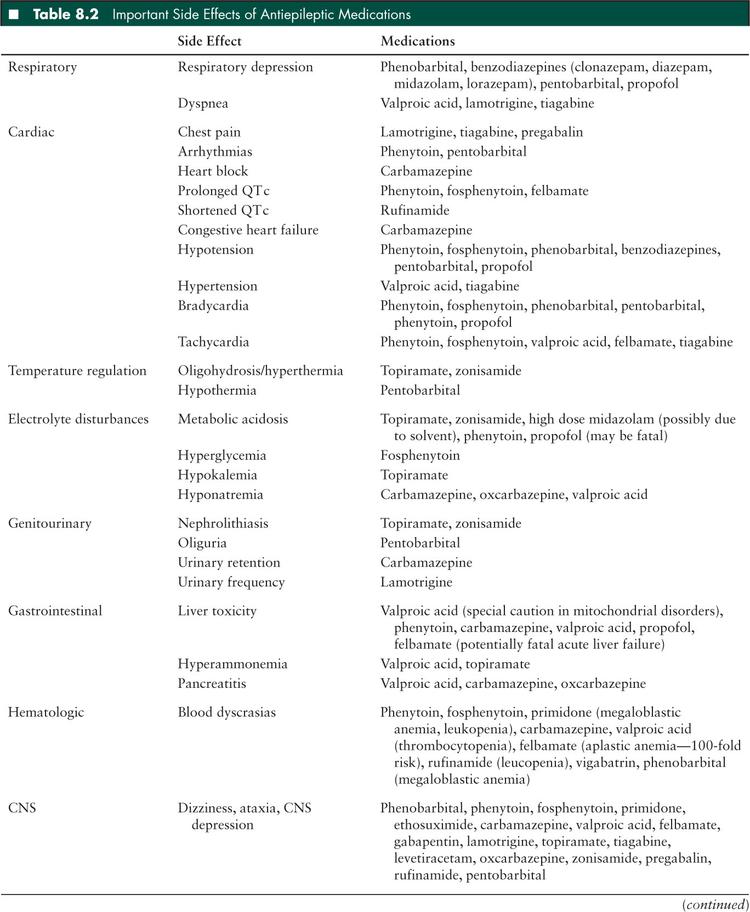
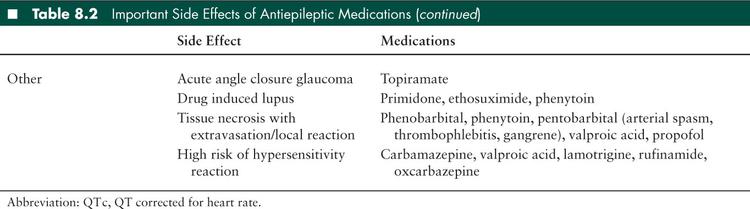
Side effects from AEDs are a common cause of morbidity in children with SE. Respiratory compromise with seizures is a common reason for admission to the PICU; Hussain et al (27) found that 13% of children admitted to the PICU with a diagnosis of seizures required ventilator support. Many of the medications, particularly the first-line benzodiazepines and phenobarbital, are respiratory depressants. In addition, many of the medications, especially when given quickly, cause hypotension which further compromises the child’s cardiorespiratory status. Individual AEDs have unique side effects which should be taken to account when choosing an agent and selecting monitoring parameters (Table 8.2). Many AEDs are processed through the hepatic p450 system and may interact with other medications that the patient is receiving; careful attention should be paid to dosing of medications in patients with either liver or renal failure.
Propofol and the risk of fatality with its use deserves special attention. Although propofol is commonly used in the adult ICUs for sedation and burst suppression, children are at high risk for developing propofol infusion syndrome. This syndrome is characterized by rhabdomyolysis, hyperkalemia, liver, cardiac, and renal failure, and metabolic acidosis (118). Alternative agents should be considered for control of SE; if the medication is used, then children should be carefully monitored for signs and symptoms of propofol infusion syndrome with discontinuation of the drug immediately if they develop.
In some children, valproate may lead to hyperammonia and hepatic encephalopathy. The increase in serum ammonium level is due to several mechanisms, the most important one appearing to be the inhibition of carbamoyl phosphate synthetase I, the enzyme that begins the urea cycle. In these susceptible patients, carnitine can avoid and treat this complication (118).
In children with refractory SE, prolonged sedation/coma compromises hemodynamic function and respiratory drive but the period of immobility may also have complications. The risk of deep venous thrombosis and pulmonary embolism should be considered. Infections, especially pneumonia or tracheitis, are common.
■ MORTALITY RELATED TO SE
Since Louis Calmeil’s early clinical description differentiating SE (état de mal) from brief seizures in 1824, SE has been viewed as a significant source of mortality. However, short-term mortality (death within the first 30 days of SE) in the pediatric population is much lower than in adults (7,119). Most studies in children report an incidence of short-term mortality of less than 10% (7,14,17,20,21,28,120–124). A systematic review found that short-term mortality in children is approximately 2.7% to 5.2% though the incidence is slightly higher in the PICU (125). Usually, mortality during hospitalization or within 30 days is due to comorbidities and the underlying disease process that led to seizures rather than a direct effect of the seizures. A few rare cases are reported in the studies in which patients died from SE (0%–2%) but the cases included children whose parents elected to defer or limit medical intervention, such as intubation.
■ LONG-TERM SEQUELAE OF SE
In a landmark study of children with SE, Aicardi and Chevrie (126) wrote that “the prognosis of symptomatic status is uniformly poor, whatever the age.” Overall, Aicardi reported that 11% of patients died during their first episode of SE and 50% of these deaths were related to the seizures. Of survivors, 37% developed permanent neurologic signs and 48% had mental retardation. Of note, Aicardi defined SE as seizures lasting for more than an hour rather than 30 minutes which may have affected outcome. However, a number of studies in the last decade have suggested that the outcome of SE in children may be less dire than previously thought.
At least 50% of children with one episode of SE will have recurrent prolonged seizures even while on prophylactic AEDs. The incidence of recurrence varies in studies, likely due to variability in inclusion criteria and time to follow-up, with incidences ranging from 3% to 56% (5,7,14,15,23,24,125,127–129). Most children who will have recurrence of SE will do so within the first 2 years (24,125). Risk factors for repeated episodes of SE include young age, progressive encephalopathy, and remote symptomatic etiology. Children with preexisting neurologic abnormalities are almost 24 times more likely to have recurrent status than previously healthy children (7,125,128). Certain epilepsy syndromes also are associated with an increased risk of prolonged seizures. Eighty-one percent of children with Panayiotopoulos syndrome and approximately two-thirds of children with severe myoclonic epilepsy of infancy (Dravet syndrome) will have at least 1 more episode of SE (15).
Children with a history of SE are at increased risk of developing epilepsy. The incidence of subsequent diagnosis of epilepsy in patients varies in studies from 13% to 74%, which is most likely secondary to differences in sample selection, length of follow-up after SE, and whether patients with febrile status were included (14,28,122, 25,130). However, the incidence of subsequent epilepsy likely reflects underlying cause for the SE because duration of the first unprovoked seizures does not increase the risk of developing subsequent epilepsy (131–133). In patients with a history of SE, including adults, the risk of developing subsequent epilepsy is highest in patients with anoxia (risk ratio of 19.9) or with structural lesions (risk ratio of 7.1) (130). Among children only, patients with focal SE are the most likely to go on to develop epilepsy, which may be partially due to the fact that this subgroup includes patients with structural lesions (125).
Whether SE leads to new or worsened neurologic deficits and behavioral and cognitive problems is a controversial topic. Animal studies and autopsies of patients who died after SE provide evidence of neuronal injury and cerebral atrophy after prolonged seizures (101,134). However, it is unclear whether these pathologic changes have clinical significance. Some studies conclude that SE does not convey any significant morbidity beyond that expected for the underlying disease process nor a difference between patients with SE compared to other children with childhood onset epilepsy (24). However other studies report that children often develop new neurologic deficits after SE. This discrepancy in results likely relates to the heterogeneity of length of follow-up and outcome measures used in the literature. Overall, it appears that most children return to their neurologic baseline except for those with an acute symptomatic etiology; neurologic deterioration more likely is related to the underlying disease process (14,27,28,120,135,136).
The impact of SE on cognitive outcome is more difficult to evaluate especially since well-documented baseline cognitive testing and formal cognitive testing are rare in studies. Some studies report no difference in mental abilities or intelligence quotient in children with epilepsy with and without SE (120,137,138). However, other studies report that up to 37% of children who were neurologically normal prior to SE developed mental disabilities (139). Of note, this percentage is similar to the percentage of children with SE with acute symptomatic etiologies, so the causal relationship might be with the neurologic disease rather than with the occurrence of SE. A meta-analysis in 1990 concluded that general SE has a “slight adverse effect” upon mental abilities (140). Overall, cognitive and neurologic outcome after SE needs to be addressed on an individual basis, taking into account the child’s comorbidities.
Children with a history of refractory SE from acute symptomatic etiologies require special consideration. These patients may have a significantly worse outcome; 100% of patients in one small case series of seven patients with refractory SE went on to develop intractable epilepsy and new neurologic deficits (122).
■ DIAGNOSTIC EVALUATION OF A CHILD WITH SE
In 2006, the American Academy of Neurology (AAN) and the Child Neurology Society published a practice parameter on the assessment of a child with SE (16). Only one of the recommendations is a Level B recommendation, though there are few definitive guidelines for studies. In this section, we offer some suggestions for testing in a patient that presents with SE based upon both the practice parameter and clinical practice at our institution, which is a tertiary care center with a 45-bed PICU with an independent neurocritical care consultation service.
General Guidelines
The AAN and Child Neurology Society guidelines state that obtaining a complete blood count and electrolytes is “common practice,” so this basic laboratory evaluation should be completed in all patients. An EEG also may be considered in a child with SE as it may be helpful. In most situations, we would recommend that an EEG be obtained in children with new onset SE, new onset seizures, or if there is concern for nonconvulsive seizures. The use of emergent EEG should be considered if available in situations where there is concern for nonconvulsive seizures, such as when the patients has not experienced ongoing improvement in mental status and/or in the paralyzed patient.
Febrile Child, Without History of Epilepsy
The diagnosis of febrile SE is high on the differential diagnosis for an otherwise healthy child between 6 months and 5 years of age who presents with a prolonged generalized tonic-clonic seizure. Unlike with simple febrile seizures, detailed recommendations have not been published for febrile SE. Certainly, an evaluation should be done for the source of infection/fever. According to the AAN/Child Neurology Society guidelines for SE, blood cultures and lumbar puncture are commonly done if there is a suspicion of systemic or CNS infection, but there is inadequate data to determine whether these studies should or should not be done. In children with a prolonged postictal state, encephalopathy, meningismus, age under 18 months, refractory SE, or focal findings on examination, we recommend that a lumbar puncture be strongly considered to test for bacterial as well as viral (especially HSV) infection. If the child is obtunded or has focal findings on examination, then head imaging should be considered to evaluate for a mass lesion prior to lumbar puncture. A magnetic resonance imaging (MRI) of the brain with and without contrast including imaging of the venous structures is recommended in children with persistent encephalopathy, focal findings on examination, or evidence of increased pressure (including elevated opening pressure on lumbar puncture or papilledema) to evaluate for an intracranial etiology for seizures (such as inflammation, ischemic or hemorrhagic injury, sinus venous thrombosis, or subdural empyema). An EEG is useful to help evaluate for interictal abnormalities or nonconvulsive seizures. If a systemic or CNS infection is suspected, appropriate antimicrobial drugs should not be delayed for definitive diagnosis.
If a child has prolonged hemiconvulsions, especially if recurrent, then the clinician should consider testing of SCN1a (voltage-gated sodium channels) for Dravet syndrome (141). Females (usually with a family history of epilepsy) and a Dravet-like phenotype but negative SCN1a testing also can be tested for protocadherin 19 (epilepsy and mental retardation in females) (142). If a child has fever and other signs of autonomic instability without any other infectious symptoms and the initial diagnostic evaluation is negative, then inflammatory/autoimmune etiologies should be considered. Rheumatologic disorders such as systemic lupus erythematosus can be associated with seizures (85).
Children with limbic encephalitis (eg, due to anti-N-methyl-D-aspartic acid receptor encephalitis) may present with seizures, fever, and cerebral spinal fluid leukocytosis. The leukocytosis may lead to a misdiagnosis of viral encephalitis. These children often will have other signs and symptoms of neurologic dysfunction including behavioral problems and movement disorders (143). Studies to consider testing for include (but are not limited to) erythrocyte sedimentation rate, C-reactive protein, neopterin, anti-NMDA receptor antibodies, antinuclear antibodies, double-stranded DNA antibodies, antithyroid antibodies, complements, antiphospholipid antibodies, and a paraneoplastic panel including voltage gated potassium channels.
Child With Known History of Epilepsy
As noted earlier in this chapter, children with known epilepsy, particularly those with history of SE, often will have prolonged seizures. Initial evaluation should focus on identifying provoking factors including missed AED doses, sleep deprivation, intercurrent illness, new medications that interact or interfere with AEDs, or hormonal changes. The AAN/Child Neurology Society guidelines recommend that AED levels be considered when a child with epilepsy develops SE (Level B recommendation) (16). If the child also is febrile, then the source of illness should be investigated to look for treatable causes of fever. Lumbar puncture likely may be deferred unless the child is immunosuppressed, has a prolonged postictal period, or has signs/symptoms of CNS infection. If a CNS infection is suspected, it should be treated presumptively.
In general, head imaging does not need to be done routinely. Exceptions to this rule include the following:
Patients with a history of or risk for hydrocephalus, especially those with a ventriculoperitoneal shunt, should be evaluated for worsening hydrocephalus and/or shunt malfunction. Standard practice includes a shunt series to ensure that the shunt is intact and head imaging (computed tomography [CT] or MRI) to evaluate size of the ventricles. A neurosurgical consult may be necessary, especially if the child has any other signs or symptoms of increased intracranial pressure.
Children at risk for intracranial hemorrhage, such as those with clotting disorders, on anticoagulation, with known history of a vascular malformation, neoplasms (especially known intracranial disease), recent ischemic stroke, or known sinus venous thrombosis. CT scan (and a coagulation profile) should be obtained, especially if there are signs or symptoms of increased intracranial pressure and/or new focal findings on examination.
Children with a history of ischemia with continued risk for further strokes. Children at high risk for cerebrovascular disease include those with known hypercoagulable states (including antiphospholipid syndrome), congenital heart defects, trisomy 21 (from moyamoya syndrome), or sickle cell disease. CT is inadequate for evaluating for ischemic injury; MRI of the brain with diffusion weighted image should be done if the history and examination are concerning for possible ischemic stroke.
Children who manifest clinical evidence of increased intracranial pressure such as Cushing triad (hypertension, bradycardia, and abnormal respirations).
Child Without Fever or History of Epilepsy
The AAN, Child Neurology Society, and American Epilepsy Society published guidelines for evaluation of the first nonfebrile seizure in children in 2000 (144). This guideline recommends that an EEG be done and states that MRI of the brain is the preferred imaging modality if imaging is needed. If a child has prolonged Todd paralysis or is not returning to baseline within several hours, then emergent imaging of the brain should be obtained. The guidelines suggest that the clinician strongly consider nonurgent MRI imaging in children with “a significant cognitive or motor impairment of unknown etiology, unexplained abnormalities on neurologic examination, a seizure of partial (focal) onset with or without secondary generalization, an EEG that does not represent a benign partial epilepsy or childhood or primary generalized epilepsy, or in children under 1 year of age.” Lumbar puncture is not routinely recommended unless there is suspicion of CNS infection. Toxicology testing and labs can be sent on a case by case basis. The AAN/Child Neurology Society guidelines on SE concur that toxicology testing is a reasonable consideration, though targeted testing is more likely to be helpful than a urine drug screen. There are no other formal guidelines published. A careful neurologic history review of symptoms and family history and physical examination (that includes ophthalmologic examination and evaluation of the skin, including with a Wood lamp) can help direct further testing. For a child without preexisting neurologic deficits, with relatively normal development and history, and with rapid return to baseline, baseline testing often is sufficient.
In some cases, an acute symptomatic etiology may be obvious, such as a recent history of traumatic brain injury, recent stroke, or CNS infection. If there is a clear explanation for seizures, then the baseline testing (electrolytes, EEG, MRI if not previously obtained) might be sufficient for diagnostic studies.
For a child with a known static neurologic disorder without risk of progression (remote symptomatic etiology), such as hypoxic-ischemic encephalopathy, perinatal/prenatal stroke, congenital CNS malformation, or chromosomal abnormality, an EEG, complete blood count, and chemistries should be obtained. A nonurgent MRI of the brain should be considered if the child has not been imaged previously. An emergent CT is indicated if there is a new focal neurologic finding or signs/symptoms of increased intracranial pressure, the child is unstable, or a MRI is not planned or feasible in the near future.
In a previously healthy child, especially infants, with an unrevealing MRI and continued encephalopathy despite control of seizures, the clinician should strongly consider evaluation for CNS infection, especially HSV encephalitis, even in the absence of meningismus or fever. In addition, in the presence of ongoing encephalopathy, an autoimmune encephalopathy should be considered and evaluated as described above.
The AAN/Child Neurology Society states that a clinician may consider testing for inborn errors of metabolism in children with SE when there is clinical indications or initial studies are negative (Class C evidence). A reasonable approach would be to pursue testing for inborn errors of metabolic disorders, specific genetic disorders, and/or mitochondrial testing if the EEG, head imaging, history, and examination do not yield a clear diagnosis and the child has a history of significant developmental delays or other unexplained neurologic problems; alternatively a clinician may elect to start the evaluation immediately if the patient has a history strongly suggestive of a metabolic or genetic problem (especially recurrent episodes of encephalopathy and emesis) or very young age (< 6–9 months). A reasonable first round of testing would be general screening tests for amino acidopathies, urine organic acidopathies, disorders of fatty acid oxidation, and mitochondrial disorders (serum amino acids, urine organic acids, ammonia, urine dip for ketones and reducing substances, liver function testing, acylcarnitine profile, carnitine profile, lactate-pyruvate ratio—serum and also spinal fluid if obtained, Creatine kinase). The clinician should verify that infants had a negative newborn screen if they were born in the United States. A microarray may be helpful as well, particularly if the child has any other dysmorphic features or chronic medical problems in other organ systems. For these children, we often will obtain a magnetic resonance (MR) spectroscopy when the initial MRI is done to avoid repeating head imaging (and associated sedation) unnecessarily. A dilated ophthalmologic examination may yield clues to a diagnosis, such as mitochondrial disease, storage disorders, etc and guide further testing if initial screening studies are negative. A reasonable approach is to approach testing in a stepwise manner after discharge from the hospital. We have been more aggressive with inpatient testing in young infants, explosive onset of epilepsy, or refractory SE without obvious cause.
Mitochondrial disorders deserve special consideration. Even if the lactic acid to pyruvate ratio is normal, testing for a mitochondrial disorder should be strongly considered in children with risk factors including: personal history or family history of sensorineural hearing loss, optic atrophy, short stature/failure to thrive, cardiac disease (especially cardiomyopathy or arrhythmias), diabetes mellitus type I, exercise intolerance, liver disease, ataxia, epilepsia partialis continua, and migraines. If several of these indicators are found in the history (or if characteristic changes are seen on the MRI), then clinicians can consider performing a dilated ophthalmologic examination, echocardiogram, and electrocardiograph for further evidence of mitochondrial disorders. In a child with history that strongly suggests a mitochondrial disorder, early testing with serum studies, such as the mitochondrial common point mutation and deletion panel, and testing for POL-G mutations is reasonable.
A large number of other genetic disorders, such as Rett, aristaless related homeobox, and cyclin-dependent kinase-like 5, may cause SE and developmental delay but we would not recommend routine testing; instead testing for specific genetic disorders should be tailored to the individual patient based on their history and presence/absence of characteristic phenotype.
■ DIAGNOSTIC EVALUATION OF SEIZURES FOR SELECTION PATIENT POPULATIONS IN THE ICU
Children with congenital heart defects are at high risk for seizures. These children may develop cerebrovascular disease (hemorrhagic or ischemic), diffuse hypoxic injury from arrhythmia or poor cardiac function, and electrolyte abnormalities. Emergent MRI of the brain with diffusion weight imaging and gradient echo sequences (or if contraindicated, head CT) should be strongly considered, especially in children with focal seizures, new neurologic findings on examination, or history of recent cardiac surgery or cardiac catheterization.
Children with sickle cell disease also require urgent attention when they present with new onset seizures or seizures with focal deficits because the seizures may secondary to acute stroke (ischemic or hemorrhagic) (65). Any child with sickle cell disease should have emergent head imaging. Head CT can be done first if there is clinical concern for intracranial hemorrhage but an emergent MRI, including diffusion weight imaging, gradient echo sequences, and MR arteriography is strongly recommended to evaluate for an acute ischemic stroke. MR venogram should be considered, particularly if the child has risk factors for or signs and symptoms of dehydration. If an EEG is performed, hyperventilation, which is often utilized as a standard activating procedure, is contraindicated (145).
We also suggest pursuing a more extensive initial evaluation in infants, especially if they are under 6 months of age. Nonaccidental trauma should be considered in the differential and head imaging should be ordered early on if there is high clinical suspicion based on the patient’s history or if there is evidence of bruising and/or retinal hemorrhages. For an infant with frequent seizures and normal MRI, consider an intravenous pyridoxine trial to evaluate for pyridoxine dependent seizures. In very young infants with frequent seizures and negative initial studies, the clinician should consider not only the standard metabolic testing (discussed above) but also lumbar puncture since spinal fluid may be useful in the diagnosis of neurotransmitter disorders (neurotransmitters, biopterin/neopterin panel, pyridoxal-5-phosphate level), nonketotic hyperglycinemia (requires simultaneous serum and cerebral spinal fluid amino acids testing), and GLUT-1 deficiency (cerebral spinal fluid glucose-serum glucose ratio).
Evaluation of seizures in children with known oncologic disease must take into account their risk for CNS involvement of their disease, infection (including CNS infections) due to immunosuppression, side effects of their medication, risk of intracranial hemorrhage (from thrombocytopenia), and hypercoagulable state. Head CT should be obtained emergently if the child is coagulopathic or has sudden deterioration in mental status. Ultimately, MRI of the brain with and without contrast should be done; imaging of the venous structures is recommended if the child has been dehydrated or recently received PEG-asparginase. In the absence of contraindications, clinicians should have a low threshold to do a lumbar puncture for evaluation of CNS infection if the child is currently immunosuppressed; in addition to routine bacterial testing, testing should include cytology, human herpesvirus 6, HSV, fungal culture, John Cunningham (JC) virus, and cryptococcus. Medication toxicity from chemotherapy, especially methotrexate, may cause seizures so the medication list should be carefully reviewed. If the child has an elevated blood pressure (especially if on high dose steroids, cyclosporine, or tacrolimus), hypertensive encephalopathy is a common diagnosis; posterior reversible encephalopathy will have characteristic changes on MRI. Other symptoms that support this diagnosis are encephalopathy, visual problems, and headache. Evaluation of immunosuppressed children for nononcologic reasons similarly should address the risk of infection and medication side effects.
Children with known CNS infection may present with seizures after admission. Electrolyte abnormalities, especially hyponatremia, may be evident on baseline labs. However, if neuroimaging has not been obtained, we recommend obtaining a MRI of the brain with and without contrast, including evaluation of the venous and arterial structures. Emergent CT should be performed if there are symptoms or signs of increased intracranial pressure.
As mentioned previously, children with global anoxia/hypoxia and those with traumatic brain injury are at high risk for seizures. If clinically indicated, evaluation for increased intracranial pressure should be done due to the risk of cerebral edema. For children admitted with sudden cardiorespiratory failure without a clear etiology, though, testing for inborn errors of metabolism is reasonable using the approach described in the preceding section.
Children on ECMO are at risk for electrolyte abnormalities, cardioembolic events, and hemorrhage in addition to hypoxic injury. Electrolytes and coagulation profile should be checked. If feasible, head ultrasound (infants) and/or portable head CT are useful studies to evaluate for large hemorrhages, though ischemic injury cannot be assessed by an early CT scan.
In all situations, maintaining cardiorespiratory stability is the most important goal, so suggested imaging, especially MRI, may need to be deferred if the child is unstable. If the child cannot tolerate sedation or a prolonged scan but there is concern for acute ischemia, then we recommend that the clinician discuss appropriate plans with the neuroradiologists since a limited MRI scan can be obtained (T2 sequence, diffusion weighted images, and apparent diffusion map).
Overall, these broad recommendations reflect our clinical practice and experience and are meant to help guide initial evaluation but ultimately evaluation for the underlying etiology should be tailored in each case to the individual patient.
■ CONCLUSIONS
Seizures and SE are common neurologic emergencies which may lead to PICU admission or occur in the context of other critical illnesses. These may occur in children with epilepsy, but generally should be considered as symptomatic of an underlying acute encephalopathy until proven otherwise. Care involves simultaneous monitoring and support of systemic functions including airways, breathing, and circulation, intervention with appropriate doses of rapidly administered AEDs, and evaluation to identify the underlying etiology.
■ REFERENCES
1. , . The history of status epilepticus and its treatment. Epilepsia. 2009;50(suppl 3):56–68.
2. , , . It’s time to revise the definition of status epilepticus. Epilepsia. 1999;40(1):120–122.
3. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1981;22(4):489–501.
4. et al. How long do new-onset seizures in children last? Ann Neurol. 2001;49(5):659–664.
5. , , . Outcome of childhood status epilepticus and lengthy febrile convulsions: findings of national cohort study. BMJ. 1993;307(6898):225–228.
6. et al. Incidence of status epilepticus in Rochester, Minnesota, 1965-1984. Neurology. 1998;50(3):735–741.
7. et al. Status epilepticus in children, adults, and the elderly. Epilepsia. 1992;33(suppl 4):S15–S25.
8. et al. Incidence of status epilepticus in French-speaking Switzerland: (EPISTAR). Neurology. 2000;55(5):693–697.
9. et al. Incidence and mortality of generalized convulsive status epilepticus in California. Neurology. 2002;58(7):1070–1076.
10. et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12(4):316–325.
11. et al. An epidemiological study of children with status epilepticus in Okayama, Japan. Epilepsia. 2007;48(6):1133–1137.
12. et al. Recommendation for a definition of acute symptomatic seizure. Epilepsia. 2010;51(4):671–675.
13. et al. Prospective study of new-onset seizures presenting as status epilepticus in childhood. Neurology. 2010;74(8):636–642.
14. , . Does status epilepticus in children cause developmental deterioration and exacerbation of epilepsy? J Child Neurol. 1999;14(12):787–794.
15. et al. Underlying neurologic disorders and recurrence rates of status epilepticus in childhood. Brain Dev. 2008;30(10):624–628.
16. et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2006;67(9):1542–1550.
17. , . Pediatric status epilepticus: a perspective from Saudi Arabia. Pediatr Neurol. 1999;20(5):364–369.
18. , , . Acute symptomatic seizure disorders in young children—a population study in southern Taiwan. Epilepsia. 1998;39(9):960–964.
19. et al. The incidence, aetiology and outcome of acute seizures in children admitted to a rural Kenyan district hospital. BMC Pediatr. 2008;8:5.
20. , . Etiology and mortality of status epilepticus in children. A recent update. Arch Neurol. 1989;46(1):74–76.
21. et al. In whom does status epilepticus occur: age-related differences in children. Epilepsia. 1997;38(8):907–914.
22. et al. Status epilepticus in children with newly diagnosed epilepsy. Ann Neurol. 1999; 45(5):618–623.
23. et al. Status epilepticus in children with epilepsy: Dutch study of epilepsy in childhood. Epilepsia. 2007;48(9):1708–1715.
24. , . Status epilepticus in a population-based cohort with childhood-onset epilepsy in Finland. Ann Neurol. 2002;52(3):303–310.
25. et al. Risk factors for status epilepticus in children with symptomatic epilepsy. Neurology. 1997;49(2):533–537.
26. , , . Status epilepticus in a population-based Virginia twin sample. Epilepsia. 2004;45(2):159–165.
27. , , . Aetiology, course and outcome of children admitted to paediatric intensive care with convulsive status epilepticus: a retrospective 5-year review. Seizure. 2007;16(4):305–312.
28. , , . Features predicting adverse outcomes of status epilepticus in childhood. Hong Kong Med J. 2004;10(3):156–159.
29. et al. Admissions to a pediatric intensive care unit for status epilepticus: a 10-year experience. Crit Care Med. 1994;22(5):827–832.
30. et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology. 2000;54(2):340–345.
31. et al. Nonconvulsive seizures in the pediatric intensive care unit: etiology, EEG, and brain imaging findings. Epilepsia. 2006;47(9):1510–1518.
32. et al. Non-convulsive seizures are common in critically ill children. Neurology. 2011;76(12):1071–1077.
33. et al. Frequency and predictors of nonconvulsive seizures during continuous electroencephalographic monitoring in critically ill children. Arch Neurol. 2006;63(12):1750–1755.
34. et al. Intracortical electroencephalography in acute brain injury. Ann Neurol. 2009;66(3):366–377.
35. , . Long-term neurological complications after hypoxic-ischemic encephalopathy. Semin Neurol. 2006;26(4):422–431.
36. et al. Electroencephalographic monitoring during hypothermia after pediatric cardiac arrest. Neurology. 2009;72(22):1931–1940.
37. et al. Seizures as a predictor of long-term neurodevelopmental outcome in survivors of neonatal extracorporeal membrane oxygenation (ECMO). J Child Neurol. 2004;19(12):930–934.
38. , . Results of extracorporeal membrane oxygenation in children with sepsis. The Extracorporeal Life Support Organization. Ann Thorac Surg. 1997;63(3):756–761.
39. et al. Seizures after head trauma: a population study. Neurology. 1980;30(7 pt 1):683–689.
40. et al. Factors influencing posttraumatic seizures in children. Neurosurgery. 1988;22(5):864–867.
41. et al. Clinical predictors of post-traumatic seizures in children with head trauma. Ann Emerg Med. 1993;22(7):1114–1118.
42. , , . Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia. 1999;40(5):584–589.
43. , , . Predictors of posttraumatic convulsions in head-injured children. Pediatr Neurosurg. 1999;30(3):127–131.
44. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 19. The role of anti-seizure prophylaxis following severe pediatric traumatic brain injury. Pediatr Crit Care Med. 2003;4(3 suppl):S72–S75.
45. , . Anti-epileptic drugs for preventing seizures following acute traumatic brain injury. Cochrane Database. 2001; 4: Art No CD000173.
46. , . Cerebral complications of nonaccidental head injury in childhood. Pediatr Neurol. 1998;19(2):119–128.
47. et al. Epilepsy associated with shaken baby syndrome. Childs Nerv Syst. 2008;24(2):169–172; discussion 173.
48. et al. Acute symptomatic seizures in CNS infection. Eur J Neurol. 2008;15(1):38–41.
49. , , . Status epilepticus in central nervous system infections: an experience from a developing country. Am J Med. 2008;121(7):618–623.
50. et al. Seizures complicating infantile and childhood bacterial meningitis. Pediatric Neurology. 2004;31(3):165–171.
51. et al. Seizures and other neurologic sequelae of bacterial meningitis in children. N Engl J Med. 1990;323(24):1651–1657.
52. , , . Viral encephalitis and epilepsy. Epilepsia. 2008;49(suppl 6):13–18.
53. et al. Outcome of herpes simplex encephalitis in children. J Microbiol Immunol Infect. 2007;40(1):34–38.
54. et al. Herpes simplex encephalitis in North West India. Neurol India. 2001;49(4):360–365.
55. et al. The correlation between neurological evaluations and neurological outcome in acute encephalitis: a hospital-based study. Eur J Paediatr Neurol. 2007;11(2):63–69.
56. et al. Childhood encephalitis in Sweden: etiology, clinical presentation and outcome. Eur J Paediatr Neurol. 2008;12(6):484–490.
57. et al. Neurologic complications in children hospitalized with influenza: characteristics, incidence, and risk factors. J Pediatr. 2007;150(3):306–310.
58. et al. Hospitalized children with 2009 influenza a (H1N1) infection in Shenzhen, China, November-December 2009. Pediatr Pulmonol. 2010;46(3):246–252.
59. Neurologic complications associated with novel influenza A (H1N1) virus infection in children—Dallas, Texas, May 2009. MMWR Morb Mortal Wkly Rep. 2009;58(28):773–778.
60. , , . Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. 2002;59(8):1224–1231.
61. et al. Acute disseminated encephalomyelitis. Neurology. 2007;68(16 suppl 2):S23–S36.
62. et al. Seizures are common in the acute setting of childhood stroke: a population-based study. J Child Neurol. 2009;24(1):9–12.
63. et al. Incidence of seizures in the acute phase of stroke: a population-based study. Epilepsia. 2008;49(6):974–981.
64. , , . The spectrum of epilepsy in sickle cell anemia. J Neurol Sci. 1994;123(1–2):6–10.
65. et al. Sickle cell disease: ischemia and seizures. Ann Neurol. 2005;58(2):290–302.
66. et al. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51(5):543–552.
67. et al. Early seizures in cerebral vein and dural sinus thrombosis: risk factors and role of antiepileptics. Stroke. 2008;39(4):1152–1158.
68. et al. Risk and predictors of early epileptic seizures in acute cerebral venous and sinus thrombosis. Eur J Neurol. 2006;13(8):852–856.
69. et al. prognosis of cerebral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke. 2004;35(3):664–670.
70. et al. Cerebral venous sinus thrombosis in children: risk factors, presentation, diagnosis and outcome. Brain. 2005;128(3):477–489.
71. et al. Cerebral venous sinus thrombosis in children: a multicenter cohort from the United States. J Child Neurol. 2008;23(1):26–31.
72. et al. Cerebral sinovenous thrombosis in children. N Engl J Med. 2001;345(6):417–423.
73. et al. Cerebral sinovenous thrombosis in children: clinical presentation and extension, localization and recanalization of thrombosis. Eur J Paediatr Neurol. 2010;14(1):80–85.
74. et al. The relationship of postoperative electrographic seizures to neurodevelopmental outcome at 1 year of age after neonatal and infant cardiac surgery. J Thorac Cardiovasc Surg. 2006;131(1):181–189.
75. et al. Risk of seizures in survivors of newborn heart surgery using deep hypothermic circulatory arrest. Pediatrics. 2003;111(3):592–601.
76. et al. Relation of seizures after cardiac surgery in early infancy to neurodevelopmental outcome. Boston Circulatory Arrest Study Group. Circulation. 1998;97(8):773–779.
77. et al. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205–210.
78. et al. Reversible posterior leukoencephalopathy syndrome in childhood: report of nine cases and review of the literature. Neurol Sci. 2010;31(2):125–131.
79. , , . Clinical utility of a continuous intravenous infusion of valproic acid in pediatric patients. Pharmacotherapy. 2007;27:519–525.
80. , , . Status epilepticus secondary to hypertensive encephalopathy as the presenting manifestation of Guillain-Barre syndrome. Pediatr Emerg Care. 2007;23(9):659–661.
81. , , . Role of intravenous levetiracetam in acute seizure management of children. Pediatr Neurol. 2009;41:37–39.
82. , , . Propofol and thiopental for refractory status epilepticus in children. Neurology. 2005;65:591–592.
83. , , . Propofol infusion syndrome in patients with refractory status epileptics: an 11-year clinical experience. Crit Care Med. 2009;37:3024–3030.
84. , , . Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Arch Neurol. 2004;61(8):1254–1259.
85. et al. Valproate-induced hyperammonemic encephalopathy. Acta Neurol Scand. 2006;114(1):1–7.
86. et al. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(suppl 5):113–117.
87. , . Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience. 2006;137(1):301–308.
88. , , . The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22(6):797–803.
89. et al. Inflammatory events in hippocampal slice cultures prime neuronal susceptibility to excitotoxic injury: a crucial role of P2X7 receptor-mediated IL-1beta release. J Neurochem. 2008;106(1):271–280.
90. et al. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(suppl 2):24–32.
91. et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142–160.
92. et al. Basic mechanisms of status epilepticus due to infection and inflammation. Epilepsia. 2009;50(suppl 12):56–57.
93. , . Myoglobinuria following unilateral status epilepticus and ipsilateral rhabdomyolysis: a clinicopathological report. N Engl J Med. 1965;272:834–837.
94. , , . Myoglobinuria and renal failure after status epilepticus. Neurology. 1978;28(2):200–201.
95. et al. Rhabdomyolysis and acute renal failure due to status epilepticus. Clin Nephrol. 1998;50(3):204.
96. et al. Recurrent reversible rhabdomyolysis associated with hyperthermia and status epilepticus. Acta Paediatr. 1995;84(9):1083–1085.
97. , , . Generalized tonic-clonic status epilepticus: causes, treatment, complications and predictors of case fatality. J Neurol. 1998;245(10):640–646.
98. et al. Severe, self-limiting lactic acidosis and rhabdomyolysis accompanying convulsions. Postgrad Med J. 1989;65(763):321–322.
99. , . Autonomic alterations and cardiac changes in epilepsy. Epilepsia. 2010;51(5):725–737.
100. et al. Early post-convulsive prolongation of QT time in children. Acta Paediatr. 2005;94(9):1243–1247.
101. . The biochemical basis and pathophysiology of status epilepticus. Neurology. 1990;40(5 suppl 2):13–23.
102. , . Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol. 1973;28(1):10–17.
103. et al. Hemodynamic monitoring prior to and at the time of death in status epilepticus. Epilepsy Res. 1998;31(3):199–209.
104. et al. Status epilepticus induces cardiac myofilament damage and increased susceptibility to arrhythmias in rats. Am J Physiol Heart Circ Physiol. 2009;297(6):H2120–H2127.
105. et al. Neurogenic stunned myocardium associated with status epileptics and postictal catecholamine surge. Intern Med. 2008;47(4):269–273.
106. et al. Cardiac pathology in status epilepticus. Ann Neurol. 2005;58(6):954–957.
107. , . Abnormalities in cardiac and respiratory function observed during seizures in childhood. Dev Med Child Neurol. 2005;47(1):4–9.
108. et al. How common is ictal hypoxemia and bradycardia in children with partial complex and generalized convulsive seizures? Epilepsia. 2010;51(7):1219–1224.
109. et al. Hypoxaemia and cardiorespiratory changes during epileptic seizures in young children. Dev Med Child Neurol. 1996;38(6):511–522.
110. , , . Acute neurogenic pulmonary oedema following generalized tonic clonic seizure. A case report and a review of the literature. Eur Heart J. 1988;9(8):933–936.
111. , , . Sudden unexplained death in epilepsy (SUDEP) following previous seizure-related pulmonary oedema: case report and review of possible preventative treatment. Seizure. 2002;11(7):446–448.
112. et al. Epileptic brain damage: the role of systemic factors that modify cerebral energy metabolism. Brain. 1978;101(4):687–700.
113. et al. Prolonged focal cerebral edema associated with partial status epilepticus. Epilepsia. 1985;26(4):334–339.
114. et al. Cerebral vasomotor reactivity in epilepsy patients. J Neurol. 2010;257(5):833–838.
115. , , . Changes in blood-brain permeability during pharmacologically induced convulsions. Prog Brain Res. 1968;29:367–384.
116. et al. Temporary alteration of cerebrovascular permeability to plasma protein during drug-induced seizures. Am J Physiol. 1972;223(2):268–277.
117. et al. MRI abnormalities associated with partial status epilepticus. Neurology. 1999;52(5):1021–1027.
118. et al. Incidence of propofol-related infusion syndrome in critically ill adults: a prospective, multicenter study. Crit Care. 2009;13(5):R169.
119. et al. Short-term mortality after a first episode of status epilepticus. Epilepsia. 1997;38(12):1344–1349.
120. . Status epilepticus in children: etiology, clinical features, and outcome. J Child Neurol. 1988;3(3):167–173.
121. . Status epilepticus in infancy and childhood. Neurol Clin. 1990;8(3):647–657.
122. et al. Prolonged treatment for acute symptomatic refractory status epilepticus: outcome in children. Neurology. 2003;61(3):398–401.
123. , . In-hospital mortality of generalized convulsive status epilepticus: a large US sample. Neurology. 2007;69(9):886–893.
124. et al. Risk factors of status epilepticus in children. Pediatr Int. 2003;45(4):429–434.
125. et al. Outcome of paediatric convulsive status epilepticus: a systematic review. Lancet Neurol. 2006;5(9):769–779.
126. , . Convulsive status epilepticus in infants and children. A study of 239 cases. Epilepsia. 1970;11(2):187–197.
127. et al. Status epilepticus after the initial diagnosis of epilepsy in children. Neurology. 2004;63(6):1027–1034.
128. et al. Recurrent status epilepticus in children. Ann Neurol. 1992;31(6):598–604.
129. et al. Recurrence of afebrile status epilepticus in a population-based study in Rochester, Minnesota. Neurology. 2007;69(1):73–78.
130. et al. Risk of unprovoked seizure after acute symptomatic seizure: effect of status epilepticus. Ann Neurol. 1998;44(6):908–912.
131. et al. Seizure recurrence after a first unprovoked seizure. N Engl J Med. 1982;307(9):522–528.
132. et al. The risk of seizure recurrence after a first unprovoked afebrile seizure in childhood: an extended follow-up. Pediatrics. 1996;98(2 pt 1):216–225.
133. , . The risk of seizure recurrence following a first unprovoked seizure: a quantitative review. Neurology. 1991;41(7):965–972.
134. , . Neuropathology of status epilepticus in humans. Adv Neurol. 1983;34:129–139.
135. et al. Outcome of severe refractory status epilepticus in children. Epilepsia. 2001;42(11):1461–1467.
136. et al. Low morbidity and mortality of status epilepticus in children. Pediatrics. 1989;83(3):323–331.
137. , , . Do seizures in children cause intellectual deterioration? N Engl J Med. 1986;314(17):1085–1088.
138. , . Febrile seizures and later intellectual performance. Arch Neurol. 1978;35(1):17–21.
139. et al. Status epilepticus in childhood: a retrospective study of initial convulsive status and subsequent epilepsies. Folia Psychiatr Neurol Jpn. 1979;33(3):337–344.
140. , . Intellectual impairment as an outcome of status epilepticus. Neurology. 1990;40(5 suppl 2):23–27.
141. et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(pt 3):843–852.
142. et al. Epilepsy and mental retardation limited to females: an under-recognized disorder. Brain. 2008;131(pt 4):918–927.
143. et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091–1098.
144. et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55(5):616–623.
145. . Electroencephalography hyperventilation and stroke in children with sickle cell disease. Clin EEG Neurosci. 2006;37(3):190–192.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


