Chapter 598 Spinal Cord Disorders
598.1 Tethered Cord
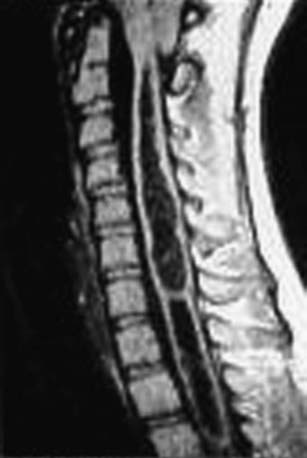
In its simplest form the tethered cord syndrome results from a thickened filum terminale, which normally extends as a thin, very mobile structure from the tip of the conus to the sacrococcygeal region where it attaches. When this structure is thickened and shortened, the conus is found to end at levels below L2. This stretching between 2 points is likely to cause symptoms later in life. Fatty infiltration is often seen in the thickened filum (Fig. 598-1).
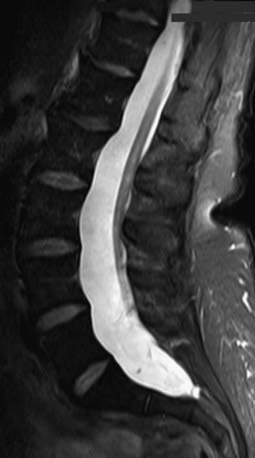
Figure 598-1 Sagittal MRI showing thickening of the filum terminale in a patient with a symptomatic tethered spinal cord.
(Used with permission from Barrow Neurological Institute.)
Any condition that fixes the spinal cord can be the cause of the tethered cord syndrome. Conditions that are well established to cause symptomatic tethering include various forms of occult dysraphism such as lipomyelomeningocele, myelocystocele, and diastematomyelia. These conditions are associated with cutaneous manifestations such as midline lipomas often with asymmetry of the gluteal fold (Fig. 598-2), and hairy patches called hypertrichosis (Fig. 598-3). Probably the most common type of symptomatic tethered cord involves patients who had previously undergone closure of an open myelomeningocele and later become symptomatic with pain or neurologic deterioration. Tethered cord syndrome can also be associated with attachment of the spinal cord in patients who undergo surgical procedures that disrupt the pial surface of the spinal cord.
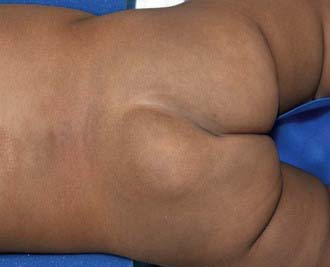
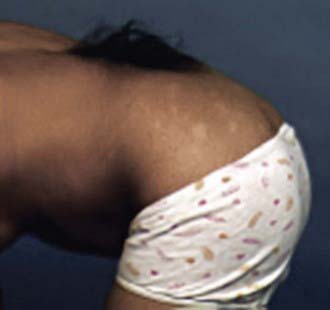
Figure 598-3 Hairy patch or hypertrichosis usually associated with diastematomyelia.
(Used with permission from Barrow Neurological Institute.)
Clinical Manifestations
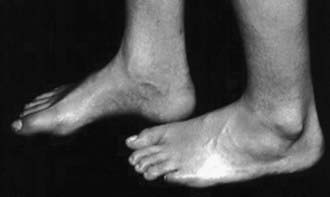
Patients at risk for the subsequent development of the tethered cord syndrome can often be identified at birth by the presence of an open myelomeningocele or by cutaneous manifestations of dysraphism. It is important to examine the back of the newborn for cutaneous midline lesions (lipoma, dermal sinus, tail, hair patch, hemangioma, port-wine stain) that may signal an underlying form of occult dysraphism. Dermal sinuses are usually located above the gluteal fold. Cutaneous abnormalities are not found in patients with an isolated thickened filum terminale. Patients who become symptomatic later in life often exhibit an asymmetry of the feet (i.e., one is smaller than the other). The smaller foot will show a high arch and clawing of the toes (Fig. 598-4). Characteristically, there is no ankle jerk on the involved side and the calf is atrophied. This condition is termed the neuro-orthopedic syndrome.
598.2 Diastematomyelia
Diastematomyelia: Split Cord Malformation
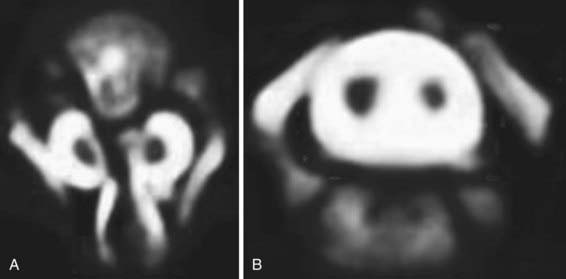
Diastematomyelia is a relatively rare form of occult dysraphism in which the spinal cord is divided into 2 halves. In type 1 split cord malformation, there are 2 spinal cords, each in its own dural tube and separated by a spicule of bone and cartilage (Fig. 598-5A). In a type 2 split cord malformation, the 2 spinal cords are enclosed in a single dural sac with a fibrous septum between the 2 spinal segments (Fig. 598-5B). In both cases the anatomy of the outer half of the spinal cord is essentially normal while the medial half is extremely underdeveloped. Undeveloped nerve roots and dentate ligaments terminate medially into the medial dural tube in type 1 cases and terminate in the membranous septum in type 2 cases. Both types have an associated defect in the bony spinal segment. In the case of type 2 lesions, this defect can be quite subtle.
598.3 Syringomyelia
Diagnostic Evaluation
MRI is the radiologic study of choice (Figs. 598-6 and 598-7). The study should include the entire spine and should include gadolinium-enhanced sequences. Specific attention should be paid to the craniovertebral junction due to the frequent association of syringomyelia with Chiari I and II malformations. Obstruction to the flow of CSF from the 4th ventricle can cause syringomyelia; therefore, most patients also should undergo imaging of the brain.