CHAPTER 13 Sexual differentiation
normal and abnormal
Normal Embryological Development of the Reproductive System
Although chromosomal sex is determined at the time of fertilization, gonadal sex results from the differentiation of the indifferent undifferentiated gonad which becomes either a testis or an ovary. This begins during the fifth week of embryological development; at this time, an area of coelemic epithelium develops on the medial aspect of the urogenital ridge, and proliferation leads to the establishment of the gonadal ridge. Epithelial cords then grow into the mesenchyme (primary sex cords), and the gonad now possesses an outer cortex and an inner medulla. In XY individuals, the medulla becomes the testis and the cortex regresses. In embryos with an XX complement, the cortex differentiates to become an ovary and the medulla regresses. The primordial germ cells develop by the fourth week in the endodermal cells of the yolk sac, and during the fifth week, they migrate along the dorsal mesentery of the hindgut to the gonadal ridges, eventually becoming incorporated into the mesenchyme and the primary sex cords by the end of the sixth week (Figure 13.1).
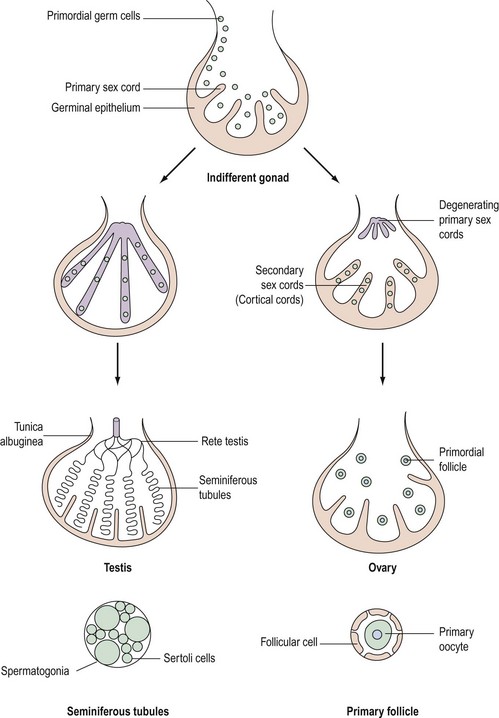
Development of the testis
The primary sex cords become concentrated on the medulla of the gonad and proliferate, and their ends anastomose to form the rete testis. The sex cords become isolated by the development of a capsule called the ‘tunica albuginea’ and the developing sex cords become the seminiferous tubules. Mesenchyme grows between the tubes to separate them (Leydig cells). The seminiferous tubules are composed of two layers of cells: supporting cells (Sertoli cells) derived from the germinal epithelium, and spermatogonia derived from the primordial germ cells (Figure 13.1).
Genetic control of gonadal development (Figure 13.2)
The pathway which controls the genetic interaction which transforms intermediate mesoderm to the bipotential gonad is beginning to be understood. It is now possible to identify with certainty two genes that are associated with this process. These two genes are Wilms’ tumour gene (WT1) and FTZ1. WT1 has been found on chromosome 11, p13 and regulates DNA transcriptase (Hastie 1994, Dong et al 1997). FTZ1 produces steroidogenic factor 1 (SF-1), and this has been found to be required for differentiation to the bipotential gonad (Wilhelm et al 2007).
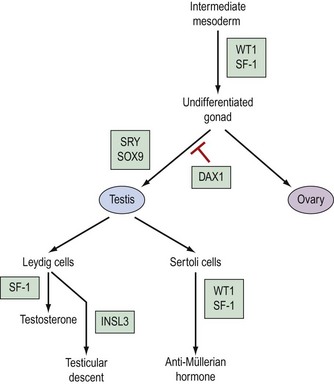
As the presence of a Y chromosome is essential for testicular differentiation, the localization of testicular-determining factor (TDF) which was localized at the short arm of the Y chromosome using cloning and DNA sequencing techniques, and the gene determining TDF was sequenced and designated SRY (sex-determining region Y gene) (Guellaen et al 1984, Behlke et al 1993). The second gene which is involved in the differentiation of the testis is known as SOX9, and this gene coexists with the SRY gene in its homeobox and is a regulator of DNA transcription. SOX9 gene expression is increased in male gonadal differentiation and decreased in female gonadal differentiation. SOX9 has also recently been shown to be a regulator of type II collagen which is involved in the formation of cartilage (Lefebvre et al 1997). The DAX1 gene is responsible for development of the receptor on the surface of the undifferentiated gonad, which allows SRY and SOX9 to differentiate the gonad to become a testis. Following the development of Leydig cells and Sertoli cells, SF-1 is involved in the control of steroid hydroxylases, and P450 aromatase causes an increase in the synthesis of testosterone and a reduction in the conversion of androgens to oestrogens, leading to a contribution to testicular regulation (Ogata 2008). In combination with ST1, it is also responsible for Sertoli cells producing anti-Müllerian hormone. Following differentiation, Leydig cells produce insulin-like 3 (INSL3) gene, expression of which leads to testicular descent and shortening of the gubernaculums (Foresta and Ferlin 2004).
The genes involved in ovarian differentiation are much more difficult to define, although DAX1 expression continues during ovarian differentiation. Therefore, it is suggested that DAX1 antagonizes the actions of the SRY gene (Swain et al 1998).
Sex chromosome anomalies
Variations may occur in the number or the structure of chromosomes in a mosaic or a non-mosaic form. The non-mosiac forms are summarized in Box 13.1.
Aneuploidy
Although there are a wide range of clinical effects, some generalizations can be made:
Structural abnormalities of X chromosomes
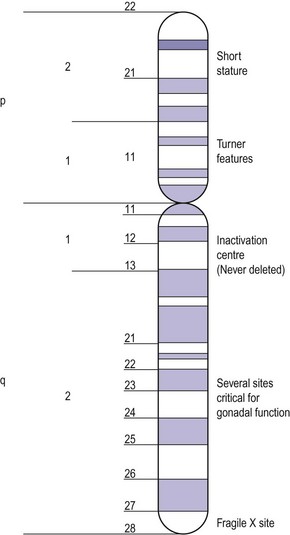
Apart from the number of X chromosomes in mosaicism, there are a number of possible variations within the X chromosome in the female. The X chromosome carries a large number of genes, not all of which have been delineated but some of which are responsible for metabolic and development disorders, and the gene map for the X chromosome is constantly being updated. If there is one normal X chromosome and the other is abnormal, inactivation of the abnormal X chromosome usually occurs in the somatic cells and this tends to diminish the effect of the abnormality, except in the ovary. Although only one X chromosome is sufficient for early ovarian development, germ cell maintenance requires several loci on the second X chromosome and on autosomes (Figure 13.3) (Simpson 1999).
Intersex Disorders
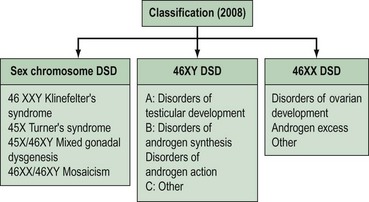
Intersex disorders are best classified into three groups (see Figure 13.4). This classification was suggested by Hughes (2008) as a new simple classification which includes all disorders of intersex.
Female (46XX) disorders of sexual development
Congenital adrenal hyperplasia
Pathophysiology
This is the most common cause of female pseudohermaphroditism and is an autosomal-recessive disorder resulting in enzyme deficiency in the biosynthesis of cortisol in the adrenal gland. Cortisol production occurs in the zona fasciculata and zona reticularis (Figure 13.5), and is controlled by adrenocorticotrophic hormone (ACTH) secreted by the pituitary gland. Adrenal androgen production occurs in the same area and is influenced by ACTH. A deficiency in any enzyme in the pathway results in decreased production of cortisol with resultant elevated levels of ACTH. This leads to increased steroid production by the adrenal reticularis and consequent hyperplasia. The stimulation by ACTH elevates the levels of circulating androgens, and this results in virilization of the female fetus.
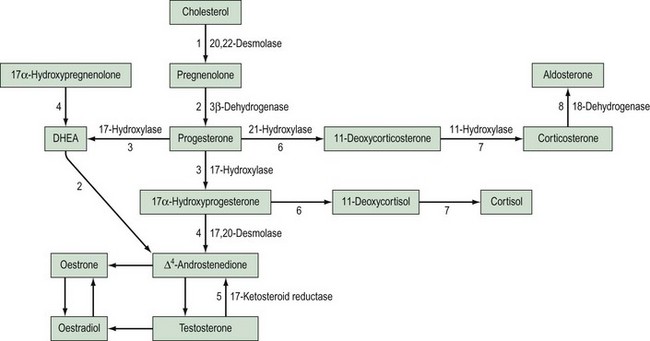
21-Hydroxylase deficiency
Aetiology
Deficiency of 21-hydroxylase is an autosomal-recessive disorder. The link between human leukocyte antigen (HLA) type and 21-hydroxylase deficiency was established by Dupont et al (1977), and this allowed mapping of the gene which was located on the short arm of chromosome 6. It is located between the HLA-B and HLA-DR loci, and subgroups of HLA-B have been closely linked to congenital adrenal hyperplasia type: HLA-BW47 is linked to salt-losing congenital adrenal hyperplasia and HLA-BW51 is linked to the simple virilizing form. Studies by Donohoue et al (1986) have shown that there are two 21-hydroxylase genes: 21-OHA and 21-OHB. Only one is active (21-OHB) and they both lie between the fourth components of complement C4A and C4B. A variety of mutations have been reported, including gene deletions of 21-OHB, gene conversions and point mutations.
Epidemiology
The incidence of 21-hydroxylase deficiency is between one in 5000 and one in 15,000, based on neonatal screening programmes (Cacciari et al 1983), although a higher incidence (one in 700) has been reported in specific populations of Eskimos (Pang et al 1982). The incidence of the non-classic form of the disease, when androgenization fails to appear before late childhood or puberty, is much more common; approximately one in 300 in the White population and one in 30 in European Jews (Pang et al 1988).
Presentation
Affected females are born with an enlarged clitoris, fused labioscrotal folds and a urogenital sinus which may become a phallic urethra. There is often great variation in the degree of masculinization of the external genitalia; this is classified according to Prader (1958).
The internal genitalia develop normally as they are not influenced by androgens.
Treatment
This is divided into four parts.
Long-term cortisol replacement therapy
Although previous reports suggested that mineralocorticoid therapy could be discontinued after infancy (Newns 1974), more recent data suggest that it is essential to continue therapy for life (Hughes et al 1979).
Surgical correction
Once the sex of rearing has been established as female, some attempt to feminize the genitalia may be made, usually within the first 18 months of life. If the clitoris is enlarged, a reduction clitoridectomy can be performed and the perineal region modified (Edmonds 1989). There are major surgical problems associated with severe virilization in congenital adrenal hyperplasia. The urogenital sinus has been formed by labial fold fusion and the folds are usually thin, but may be thick with associated narrowing of the lower vagina, especially in salt-losers. If the labial folds are thin, division by a simple posterior incision may be performed around 3 or 4 years of age. However, thick perineal tissue should be left until after puberty, and no attempt at surgery should be made until the girl is physically and mentally sexually mature. The operation, when it is performed, involves a flap vaginoplasty with a pedicle graft of labia used to recreate the vagina. Alternatively, a Williams vaginoplasty may be used.
Mulaikal et al (1987) reviewed the fertility rates in 80 women with 21-hydroxylase deficiency; of 25 with the simple form who attempted pregnancy, 15 were successful, whereas only one of the salt-losers who tried to become pregnant succeeded. There is no doubt that a major reason for the failure of the salt-losing group is the disappointing results of surgery and subsequent lack of adequate sexual function.
11-Hydroxylase deficiency
This is the hypertensive form of congenital adrenal hyperplasia which accounts for approximately 5–8% of all cases (Zachmann et al 1983). The absence of 11-hydroxylase leads to elevated levels of 11-deoxycorticosterone (DOC), and although this means a decreased amount of aldosterone, DOC has salt-retaining properties, leading to hypertension. Androstenedione levels are also elevated and this can result in ambiguous genitalia.
The genetics, however, are rather different. There is no HLA association with 11-hydroxylase deficiency, but the use of a DNA probe has located the gene on the long arm of chromosome 8 (White et al 1985).
Androgen-secreting tumours
Ovary
A number of androgen-secreting tumours have been reported, including luteoma (Hensleigh and Woodruff 1978, Cohen et al 1982), polycystic ovaries (Fayez et al 1974), mucinous cystadenoma (Novak et al 1970, Post et al 1978), arrhenoblastoma (Barkan et al 1984) and Krukenberg tumours (Connor et al 1968, Forest et al 1978). Not all female fetuses will be affected and there is no association with gestation and exposure. The fetus may be partly protected by the conversion of the maternally derived androgen to oestrogen in the placenta, and thus the degree of virilization is variable.
Adrenal gland
There are only two reports of adrenal adenomas causing fetal masculinization (Murset et al 1970, Fuller et al 1983). These tumours may be responsive to human chorionic gonadotrophin, and thus levels of androgen may be higher in pregnancy than in the non-pregnant state, leading to androgenization of the fetus.



