Fig. 10.1
Sleep and arousal centers in the human brain. (a) Artistic rendering of the human brain in the awake state illustrating important arousal and sleep centers and pathways of neurotransmission. Cholinergic input (orange) from the laterodorsal tegmental (LDT) and pedunculopontine (PPT) nuclei project through the thalamus and facilitate thalamocortical transmission of arousal signals. A second pathway projects through the hypothalamus to cortical centers and facilitates the processing of thalamocortical inputs arising from midbrain centers including the noradrenergic (blue) locus ceruleus (LC), the serotonergic (purple) dorsal raphe (Raphe), the histaminergic (pink) tuberomammillary nucleus (TMN), and the dopaminergic (yellow) ventral periaqueductal gray matter (VPAG). This pathway also receives input from the cholinergic (orange) basal forebrain (BF) and the peptidergic neurons of the lateral hypothalamus (LH) and perifornical neurons (PeF), which contain orexin or melanin-concentrating hormone (light green). The melatonergic (red) neural network affects arousal and sleep through the regulation of circadian rhythms. This internal biological clock originates in the suprachiasmatic nucleus (SCN) and projects through the dorsomedial hypothalamus (DMH) sending inhibitory signals to the GABAergic (gray) ventrolateral preoptic nucleus of the hypothalamus (VLPO). (b) Artistic rendering of the human brain in the sleeping state illustrating important sleep and arousal centers and pathways of neurotransmission. The VLPO of the hypothalamus sends descending GABAergic (gray) inhibitory signals to the midbrain arousal centers including the PeF, TMN, VPAG, Raphe, LDT and PPT, and LC. During the early hours of dark periods, the pineal gland (Pin) releases melatonin (red ), which has inhibitory effects on the SCN and DMH of the melatonergic system. Nuclei that control neural activity during rapid eye movement (REM) sleep have been identified in the pontine midbrain. The pericoeruleus (PC) and parabranchial (PB) nuclei send glutaminergic (green) projections through the BF to affect cortical activity during REM sleep, and projections from the sublaterodorsal nucleus (SLD) send glutamatergic signals through the spinal cord to induce atonia that is characteristic of REM sleep (Reprinted with permission from Wafford KA, Ebert B. Emerging anti-insomnia drugs: tackling sleeplessness and the quality of wake time. Nat Rev Drug Discov. 2008 Jun;7(6):530–40)
On the opposite side of arousal are sleep-promoting nuclei, the most important example being a nucleus in the hypothalamus called the ventrolateral preoptic nucleus (VLPO), a brain region also referred to as the “preoptic area” to indicate how close it is to the optic nerves. This nucleus is active during sleep, and actually inhibits other arousal nuclei [17]. The neurotransmitter mediating VLPO’s effects is gamma amino butyric acid (GABA), which happens to be the target of many sedatives (benzodiazepines, etomidate, propofol). However, GABA receptors are also widely distributed throughout the brain, being concentrated in certain brain regions such as the medial temporal lobe [18]. Thus GABAergic drugs act at many locations other than VLPO. GABA receptors are not only widely dispersed throughout the brain, but come in some 18 varieties, depending on which and how particular subunits making up the receptor are configured. Thus “GABAergic” drugs differ from each other depending on how they interact with receptor subspecies [19]. This will be further elaborated in the section on etomidate, which targets GABA receptors containing the alpha5 subunit [20]. GABA receptors are notably present in brain regions important in memory processing, namely medial temporal lobe structures comprising the hippocampus and amygdala [21]. These brain regions are located close by the brainstem and hypothalamus, and the collection of these structures may be considered as a functional unit upon which higher cortical centers depend. It is no surprise that one of these systems (e.g., sleep pathways) will influence function of other systems (e.g., memory). Thus, drugs acting at GABA receptors will have effects on memory as well as arousal (sedation).
The known physiology of sleep pathways is ever expanding, and increasingly complex interactions are teased out as new pathways, neurotransmitters, and nuclei are discovered. A recent example is the orexinergic pathway, so named because the transmitter in question is orexin [22]. This neurotransmitter may be familiar as a mediator of eating behaviors, and how this system relates to obesity is an active and exciting area of research [23]. Turning back to sedation and sleep, a lack of orexin leads to narcolepsy [8]. No doubt, in the future, sedative drugs targeting the orexinergic pathways will become part of our armamentarium, presumably after pharmacologic congeners have been marketed for treatment of obesity. It is fruitful to conceptualize arousal and sleep nuclei and their neurotransmitters dancing in a carefully choreographed harmony [24]. This careful seesaw balancing act allows transitions between states of sleep and arousal over short periods of time as one set of nuclei come on line and inhibit the others. The behavioral correlate of this activity is the seemingly rapid transitions between “nodding off” and startling back awake. These mechanisms may play a role in the quality of sedation experienced with dexmedetomidine as patients seem to pass through similar rapid transitions. It should be noted, however, that these transitions occur on top of a background of tonic inhibition. One is not fully awake and then suddenly asleep. Thus, nodding off occurs only when a certain level of background sedation (non-arousal) is present, such as occurs with sleep deprivation and the accumulation of melatonin in the brain. This also seems to be a characteristic when dexmedetomidine is given, and further detailed in the section on dexmedetomidine. Sleep (adequate sedation) is not possible until a given “tonic” state of sedation develops as the drug is being infused. Even then, arousal from dexmedetomidine sedation may occur quite easily compared with other sedative agents.
To Sedate, Perchance to Not Remember!
If a patient is sedated to the point that they do not respond to their surroundings, then they will also not remember what is happening in their surroundings. Implementation of “I don’t want to remember anything!” relies on this fact, and sedation is usually administered at a dose that produces unresponsiveness. Clinically this is a much easier goal to target than producing “amnesia,” where the patient is responsive, but will not subsequently remember (more on this later) [25, 26].
The heuristic of producing unresponsiveness to ablate memory formation naturally leads to the question of what is it to “experience” something? This is not a trivial question, and can certainly enter the realm of philosophy. For purposes of this chapter, conscious experience is defined in terms of brain processes [27]. Experience begins when information from the outside world registers in the brain, the initial portal being the sensory cortices via transmission through the thalamus. The thalamus is a deep-seated set of nuclei that one can consider as analogous to the key Internet hubs through which the world’s information flows [28]. But sensory “experience” is by itself not sufficient for “Experience” with a capital E [29]. Information from different parts of the brain must be integrated into what is more formally termed a “percept.” Integration occurs not only from sensory cortices, but also from memory areas that represent knowledge of the world, those being semantic memories. The integration of these memories allows the events just experienced to be deciphered as a conscious experience. After this percept forms, it has the chance to be (consciously) remembered [30, 31] (Fig. 10.2). As providers of sedation, we can interfere with any of these stages using our armamentarium of sedative potions. Our influence on these events may seem to be greater than it actually is. In fact, even when fully anesthetized, sensory experience still occurs, but as it happens that sensory input remains pretty much localized to the sensory cortices; how this happens will be explained later in this chapter [32–35]. The murky question is whether the arrival of sensory input during full anesthesia has any influence on anything afterwards—do we form unconscious memories of some sort [36, 37]? As sedation is really anesthesia using lower doses of drugs, this becomes even a more vexing issue assuming a dose–response relationship. To this point there is no literature to guide us. The state of affairs with auditory input during sedation may be analogous to the not-so-distant issue of whether neonates perceived pain, and whether this was important to prevent. When it became evident that neonates did, in fact, perceive pain, it was felt that it would be in the best interest of the patient to prevent this. Narcotics (or other methods of alleviating pain) became a requisite part of the anesthetic management of neonates during surgery. So, the philosophical question is whether children who are sedated, who no doubt receive auditory input that is registered in the sensory cortices of the brain, should also be afforded similar considerations. Fortunately for a significant percentage of pediatric patients, they have foam earplugs inserted for magnetic resonance (MR) scanning. But for others, should we not be aware of the nature of auditory input during sedation? This rhetorical question has no answer to date, but does set the groundwork for interesting research in the future. For example, at clinically relevant doses, pentobarbital is associated with auditory activation, whereas propofol does not seem to be [38].
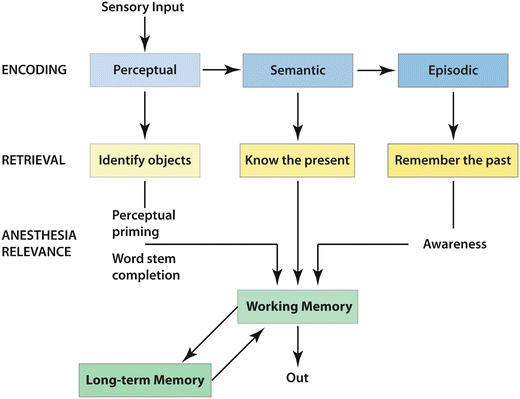
Fig. 10.2
Serial parallel independent memory (Adapted from Schacter DL, Tulving E. Memory systems 1994. Cambridge, Mass., MIT Press, 1994)
The ability to influence information processing after sensory perception using sedative agents is much greater, as processes of information integration and subsequent memory formation are much more sensitive to drug action. Consciousness is the binding of information across different brain regions into a thought, if internally generated1, or a percept generated from external sensory inputs. The percept incorporates sensory parameters (nature of the object/sound being experienced), previous knowledge (semantic memories; e.g., “This object is a red block”), and personal subjective memories of experiences (i.e., episodic memories; e.g., “I played with this block last time”) [39]. Another way to think of sedation is that it is administered so as to prevent information integration; i.e., when the child is “asleep,” they are not able to integrate information into a percept [2, 33, 40–45]. Sensory input may (and undoubtedly does) arrive in the brain, but it has nowhere to go. Processes connecting part A of the brain with part B are rendered largely nonfunctional by the sedative agent, so there is no chance for memory formation—at least of conscious memory (which is the form of memory we are most concerned with when we “don’t remember a thing”) [46, 47].
Memory: What Is It, Really?
Memory is not a unitary process, but rather a complex set of interrelated physiologic processes, and as such is continually malleable over time. At its simplest level, there are two distinct forms of memory—conscious and unconscious—and this chapter will focus on the former [48]. Conscious memory requires the formation of a percept, which is the sine qua non of consciousness. In other words, one has to be conscious to form a conscious memory. Thus, when someone is unconscious, for the purposes of this chapter specifically as a result of sedation, conscious memories will not be formed. As an illustrative side point, questions of whether patients in persistent vegetative states (PVSs) are “conscious” revolve around the question of information integration, the ability to form a percept from external or internal experience [49]. As someone in a PVS cannot respond, electrophysiologic evidence of information integration is sought in these patients. Various approaches have been used to identify electrophysiologic signatures of information integration to use as a surrogate of “consciousness.” However, even very sophisticated methods suffer from statistical artifacts, and one still cannot be sure whether consciousness is or is not present. On a much larger scale, this also is the case during sedation/anesthesia. To date, there is no reliable EEG-type monitor (or for that matter, any other type of monitor) that can tell us whether a patient is sufficiently sedated, sufficiently anesthetized, or sufficiently amnesic for a particular situation [50–53]. Thus, it is not surprising that even when we think a patient is well sedated, they can be “playing possum” and suddenly wake up; i.e., they have the ability to integrate information despite our best efforts. This fear of sudden arousal leads to even higher dosing of sedative agents, wandering dangerously close or actually into the state of anesthesia (by definition preventing arousal in 50 % of patients when a surgical incision is made). Thus, it would be desirable to utilize sedative agents that not only sedate but can also affect the last stage of memory formation, that of incorporation of the conscious percept into a lasting conscious memory. This last process is the most sensitive to drug effect, occurring at concentrations lower than those producing sedation, as long as the drug has specific amnesic properties [54–56]. These are much fewer in number than those producing sedation, but they are widely used, consisting of the benzodiazepines, propofol, and ketamine. These drugs have the ability to impair formation of long-lasting memories (defined as longer than 30–60 min) for conscious percepts formed during periods of arousal sufficient for information integration, a state often termed “awareness.”
To be aware is to experience your surroundings and can be proven by responding appropriately to verbal commands (e.g., “Squeeze my hands twice.” Parenthetically it is very difficult to prove or disprove awareness without any behavioral measures, and that is why it is difficult to know if patients in a PVS are conscious) [57, 58]. In fact, a significant proportion of patients who are “fully” anesthetized can be aware, but virtually none of them consciously remember this experience of awareness afterwards [59].
The Last Building Block of a Conscious Memory: Consolidation
This is the key section in which “amnesia” will be defined. To be clear, amnesia in this chapter is being used in the context of administration of sedative drugs [25, 60]. Amnesia generally refers to any pathologic state in which memory is affected (e.g., Alzheimer’s dementia, transient ischemic amnesia, traumatic amnesia, etc.). These amnesias can be classified as either anterograde or retrograde, depending on which memories are affected. If the insult affects only memory after the insult, this is termed anterograde amnesia and is the type of amnesia produced by drugs (more on this later). A much more intriguing type of memory loss is retrograde amnesia, subject of many movies. This refers to loss of memories before the insult, usually some type of traumatic event (e.g., head injury, electroconvulsive therapy), where the time frame of loss of memory ranges from minutes (not remembering how the accident occurred, for instance) to months, possibly years. Retrograde memory loss has never been convincingly demonstrated for any drug to date. “Amnesia” as used in this chapter (and generally any investigation of acute drug administration) refers to the drug-induced inability to remember a conscious percept and in many studies is demonstrated by the lack of memory for pictures or words seen or heard while drug was being given [56]. The presence of conscious percepts allows one to be aware, in other words able to integrate information either from external or internal sources. We can only observe external awareness, as illustrated by the question of whether patients in a PVS are internally aware. Awareness is evidenced by appropriate behaviors to the environment (e.g., ability to follow commands in the presence of sedative drugs). Awareness occurs in the “here and now,” but to remember being aware at some past point requires further processing of information into a memory. In order for this to occur, the conscious percept has to be consolidated into a lasting memory [55] (Fig. 10.3).
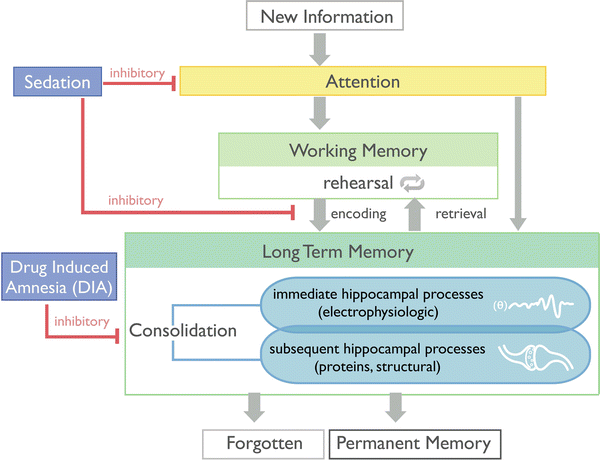
Fig. 10.3
Flow of sensory input comparing amnesia versus memory formation during sedation (Reprinted by permission © Memorial Sloan-Kettering Cancer Center)
Consolidation is a fundamental area of neuroscience investigation, finding its roots in the works of Donald O. Hebb in the middle of the last century. Hebb was a psychologist keenly interested in the basis of memory and proposed that the brain was plastic; in other words, neurons changed their connections (synapses) with each other based on how they were activated [61]. In essence, a memory resides in the altered synaptic connections in the brain [62]. Thus, consolidation produces a brain different from what it was before and that difference is a memory. These specific changes, referred to as Hebbian learning, are the result of many dozens of physiologic and molecular processes, each having its own particular time frame [63, 64]. The sum total of these processes are termed consolidation and become active once a conscious percept is learned, defined as acquiring information from the outside world. Consolidative processes are present to varying degrees for the life of the memory. A corollary of the fact that consolidative processes are continuously active is that memories are continuously malleable [65–68]. The whole question of the reliability of eyewitness testimony revolves around this physiologic fact. Other times, malleable memories can get out of hand, resulting in “flashbacks” of increasing intensity producing posttraumatic distress syndrome where memories are closely linked to flight or fight fear responses [69–71]. But the most common fate of memories is that they decay over time [72].
Just as the thalamus is a key hub for sensory experience and integration that allows consciousness to exist, a seahorse-shaped structure in the medial temporal lobes (almost adjacent to the thalamus, as it turns out) called the hippocampus is a similarly important mediator of conscious memory [73–75]. The hippocampus connects incoming information with diverse locations in the brain, where previous memories reside, to allow new memories to be created (Fig. 10.4). Without the hippocampus, no conscious memories can be formed, and this was first appreciated in the famous neurologic case involving a patient with the pseudonym HM. HM had bilateral temporal lobectomies for the treatment of intractable epilepsy in 1956 [75]. This operation was a well-accepted form of treatment in the era where the brain was considered to operate under the principle of equivalency. This principle rests on the thought that if one part of the brain was damaged or removed, another part would take over that function, thus minimizing the impact of pathology or surgical intervention. Indeed, it was thought that surgical intervention could remove the diseased focus in the brain, thus ameliorating symptoms of the disease, in particular epilepsy [76]. To a large extent this was true for epileptic foci in many cortical regions. However, epileptic foci commonly reside in the temporal lobes, and it soon became apparent that significant removal of the medial temporal lobes bilaterally (which happened to contain the hippocampi) resulted in severe impairments of conscious memories. This was described most famously by Scolville and Milner [75]. For those interested, a quite accurate depiction of what life would be like without the ability to form conscious memories is made in the film Memento [77]. The hippocampus is also important in “readout” of newly formed memories, and in a sense this readout is “practiced” during certain stages of sleep [78–81]. Sleep will improve conscious memories, and the adage “get a good night’s sleep” is not without merit [82]. Not surprisingly, detailed investigations of how hippocampal mechanisms interact with memory are ongoing topics of research.
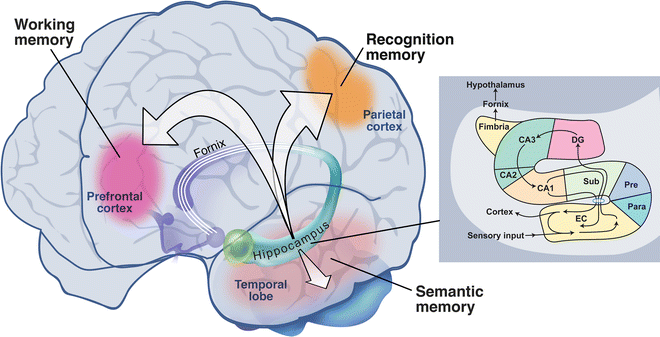
Fig. 10.4
Memory regions of the brain. Inset shows sensory pathway (Reprinted by permission © Memorial Sloan-Kettering Cancer Center)
The First Building Block of Amnesia: Forgetting
The fate of virtually all memories is that they are forgotten over time. This is a fortunate event, as otherwise our brains would be filled with useless information. Some rare cases of the inability to lose old memories have been described [83]. Ebbinghaus, a psychologist, first described the decay of memories over time over 150 years ago [84]. Most memories are forgotten soon after learning, but this process continues at a slower pace as long as we measure it [72]. They key to understanding the nature of drug-induced amnesia is to understand the forgetting of memories over time. For, in fact, memories can be and are formed in the presence of amnesic drugs [85]. The fact that at low concentrations of, for example, benzodiazepines, virtually all processes required to form a conscious memory are operational—namely, sensory perception, information integration, learning, and initial conscious memory formation—explains why behavior is visibly normal, other than possibly being affected by some sedation (e.g., being “drunk”). If we measure what happens to memories formed in the presence of an amnesic drug over time, we find that they disappear very rapidly [54]. In fact, in the case of midazolam or propofol, no discernible memory is present after 30–60 min (Fig. 10.5). Thus, drug-induced amnesia is typified by the inability of memories to be remembered over time. The mechanism(s) underlying this effect is (are) still unknown, but could be summarized as the inability of consolidation to function normally, so that the memory cannot be retained at its usual strength over time. These qualities set the stage for an event such as date rape, where a small amount of a substance active at GABAergic receptors is secretly administered, usually with alcohol to mask any sedative effect. No suspicion is aroused; observers note no particularly abnormal behavior [86]. However, the victim experiences a complete lack of even the most traumatic memories of events transpiring in the presence of the drug. Fortunately these same qualities are much more commonly used for beneficial uses, where propofol or midazolam (and likely ketamine) can produce amnesia for events transpiring in the presence of drug even when the patient is awake (or using more accurate terminology: aware).
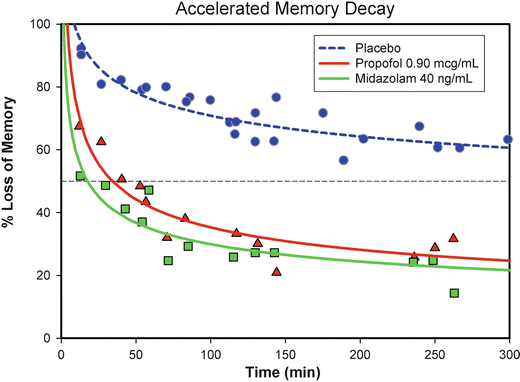
Fig. 10.5
Memory decay following propofol and midazolam compared to placebo (Reprinted by permission © Memorial Sloan-Kettering Cancer Center)
Mechanistic Implications of Drug-Induced Amnesia
Long-term memory is the final stage of processing of information acquired from the outside world, a process termed “learning.” Learned information has thus experienced sensory and cognitive manipulations located in diverse regions of the brain. Thus, a sine qua non of memory function is that different parts of the brain must be able to communicate amongst themselves. These processes of communication have become the mechanisms of most interest in terms of understanding anesthetic and sedative drug actions on cognition and consciousness [44–47, 87]. Using quite sophisticated measurement and analytic techniques, connectivity between different brain regions can be quantitated [88]. Measures of the complexity of information (information content) flowing between these regions can be obtained as well [89]. It is no surprise that such complex processes are inhibited at low doses of anesthetic sedatives. Memory impairment from sedation is based on these inhibitory mechanisms [90]. At higher drug doses, key brain regions participating in the maintenance of consciousness, namely, the thalamus, are disconnected from the rest of the brain (or, alternatively, the cortical brain becomes disconnected from itself (clustered) preventing information flow), and the person falls asleep, or more correctly is anesthetized [88, 91, 92].
Similar inhibitory mechanisms of information processing could be at play to produce amnesia when sedation is minimal. As explained previously, amnesia is best characterized as the inability to retain a memory over time, which means that a memory was formed in the first place—in other words, learning has occurred. Learning can only occur if all the processes important in the formation memory function well. Behaviorally, the person experiencing an amnesic concentration of drug will appear to be relatively normal, as most of their brain functions are working well. Specifically, information is transmitted through the thalamus to sensory cortices of the brain, and this information is forwarded to other brain regions for processing. The first way station after sensory perception is working memory, which can be considered a scratchpad containing information from different parts of the brain, which are then collated into a percept [93, 94]. These working memory processes are located in the front part of the brain (prefrontal cortex) and involve communications with the thalamus and hippocampus to process a memory [95]. Working memory processes are transient and are most sensitive to sedation [96, 97]. As sedation increases, these processes become increasingly impaired, and information cannot be then further processed and thus cannot be learned as a memory. We all experience this when we are too sleepy to remember some bit of information given to us (prototypically a telephone number, thus the popularity of napkins as external scratchpads in bars). Ergo, for the amnesic effect of a drug to be expressed, working memory must be intact. Newly acquired information must be transferred from working memory into long-term memory stores located in diffuse regions of the brain, with the hippocampus anchoring learning and retrieval of these memories [55]. The long-term memory that was just learned in the presence of amnesic drugs is then quickly forgotten. Both long-term and working memory processes can be indexed by electrophysiologic measurements. Indeed, amnesic drugs affect electrophysiologic measures of long term, but not working memory processes [85].
Memories We Don’t Know We Have: The Unconscious Mind
This chapter will focus on conscious memory processes, as described so far. This is primarily related to the fact that it is much easier to study the effects of drugs on conscious memory, as behavioral changes (i.e., recognition of previously experienced stimuli, “Did you see this picture before?”) are robustly measureable. Unconscious memories are hard to detect, as changes in behavior based on these memories can be quite subtle. It is especially difficult to determine if a change in a memory-related behavior is as a result of effects on conscious or unconscious processes. Thus, controversy usually surrounds the interpretation of these studies in terms of drug effects on unconscious versus conscious memories [37].
Clinical Practice
In clinical practice, it is unusual, particularly in pediatric patients, to produce a state where some quantity of drug is given, but at a dose where little sedation is apparent. Such a situation would amplify the differences between amnesic (e.g., propofol, midazolam, ketamine) and non-amnesic (pentobarbital, probably dexmedetomidine) drugs. Typically, in practice a deep state of sedation is produced, where little responsiveness is present. As detailed previously, if stimuli are not perceived, then they will not be remembered (at least as conscious memories). When propofol is administered in a similar fashion to dexmedetomidine, namely, giving a loading dose over a 10-min period, the effects on memory between these drugs are virtually indistinguishable, despite the fact that propofol has stronger and more specific amnesic properties. However, what is clinically relevant is if drug concentrations become low enough that patients wake up and become responsive while they still have some drug in their system. At these low concentrations, amnesic drugs such as propofol will prevent retention of any memories learned in this state, whereas dexmedetomidine will probably not.
Sedative Agents: Brief Considerations
The adjective “brief” is used to represent the fact that very few studies have been conducted to examine the interaction of drugs with sedation versus their amnesic effects. As discussed so far, both effects are closely related and are difficult to dissect out. The vast majority of studies examining the interaction of sedative drugs with memory have been conducted in healthy adult volunteers, in order to control the many factors that can affect memory other than the presence of drug. Remarkably fewer studies have been conducted in children, and to a large extent, as far as memory effects of drugs are concerned, children are considered as “small adults,” which undoubtedly is not true. However, at this time, this is all we have to guide us.
Propofol
The story of how propofol’s effects on memory were discovered mirrors the increased knowledge of potential mechanisms by which many anesthetics affect conscious memory processes. To a certain extent, this knowledge reflects the realization that anesthesia is not a unitary event affecting one neurobiologic target, but rather a blend of many effects [4]. Even for a given effect (e.g., memory impairment) probably, there are diverse targets of different anesthetics, but these likely lead to a final common pathway that is the critical effect causing amnesia. As conscious memory is dependent not only on certain neuroanatomical substrates but also distributed processes involved with information flow and communication, multiple potential targets are candidates for a critical common pathway designation. One example is the theta oscillations of brain activity associated with memory processes [98].
It is only recently that specific actions of anesthetics on memory have been generally appreciated. For many years, it was accepted that all sedative drugs were just that and any effect on memory was a “side effect” of sedation, such as one sees from alcohol and other commonly used sedative medications. As detailed above, sedation impairs memory when it is of a sufficient magnitude. A not-so-fortuitous conjunction of events initially branded propofol as a non-amnesic drug. The great advantage of propofol lies in its pharmacokinetics, such that the drug virtually disappears from the blood stream before one’s eyes [99]. Propofol rapidly migrates from the blood (and thus the brain) into the vast pharmacologic reservoirs of the body, and so its effect is very transient after a single or even multiple boluses. Because of these properties, propofol could be used to induce deep sedation, with little or no accumulation of drug. Thus a rapid wake-up resulted even after substantial doses of drug were given with little, if any, hangover effect. Propofol quickly became the drug of choice in many situations, not only for sedation but anesthesia as well. But the very same pharmacokinetics could work against the provider. When propofol was administered by intermittent bolusing rather than continuous administration, a great likelihood existed of subtherapeutic drug concentrations being present between boluses because the drug disappeared so quickly from the blood. Subtherapeutic concentrations meant that there was very little chance of amnesia being present, as amnesia occurs only over a very small dose window. Inevitably, case reports describing this exact situation were published and supported the notion that propofol could not be used to reliably prevent memory formation [100–103]. However, it was not the drug that was the issue, but the way it was administered. When propofol is administered by continuous infusion and constant blood levels are maintained, propofol is as good as an amnesic drug as any other, most notably the benzodiazepines—the yardstick of amnesic agents [26, 55, 56, 85, 104]. Fortunately, drug infusion pumps are now much more commonly available and are routinely used to administer intravenous medications, which continue to evolve to be ever more short acting.
Propofol was an ideal agent to use in volunteer studies to dissect out how anesthetics might impact conscious memory because of the rapid onset and offset pharmacokinetics and, not insignificantly, its anti-nausea properties [105]. Initial studies focused on carefully measuring sedation, and when these were equated amongst different drugs, memory impairment for events occurring in the presence of drug differed substantially between amnesic and sedative drugs, namely, propofol and midazolam versus thiopental or fentanyl [56]. These observations indicated that two separable drug effects on memory were present and laid the groundwork to further delineate properties of drug-induced amnesia using propofol as a prototypical amnesic agent.
The next key observation was that propofol (and midazolam) did not prevent formation of memories, other than some mild impairment from associated sedation [54, 85, 106]. A most useful conceptualization of memory processing was that of memory being the end result of the flow of information from the outside world, through transient working memory processes into a final conscious memory (Fig. 10.3). Sedation affected initial working memory processes (just as divided attention does by diverting resources away from working memory) and indeed would prevent memory formation. However, the amnesic effect of propofol (and midazolam) occurred after the memory was formed, in other words during the consolidation process.
Further studies refined the nature of this amnesic effect. The loss of information over time could be modeled using a power decay curve, and the rapid loss of memories formed in the presence of amnesic drugs was reflected in the rapid decay constant of this curve. Initial strength of the memory was separable in another parameter and reflected the sedative effects of the drug in question (Fig. 10.6). These measures parameterized previous observations, namely, that sedation prevented memory formation (decreasing initial strength of the memory), whereas amnesic actions prevented the consolidation of any memories that were formed or, in other words, increased the rate of forgetting. In the case of propofol and midazolam, memory was lost within 30–60 min after being formed. A key question is: When exactly in the consolidation time frame do these drugs produce amnesia? In a sense the answer can be “back engineered” based on the following thought experiment.
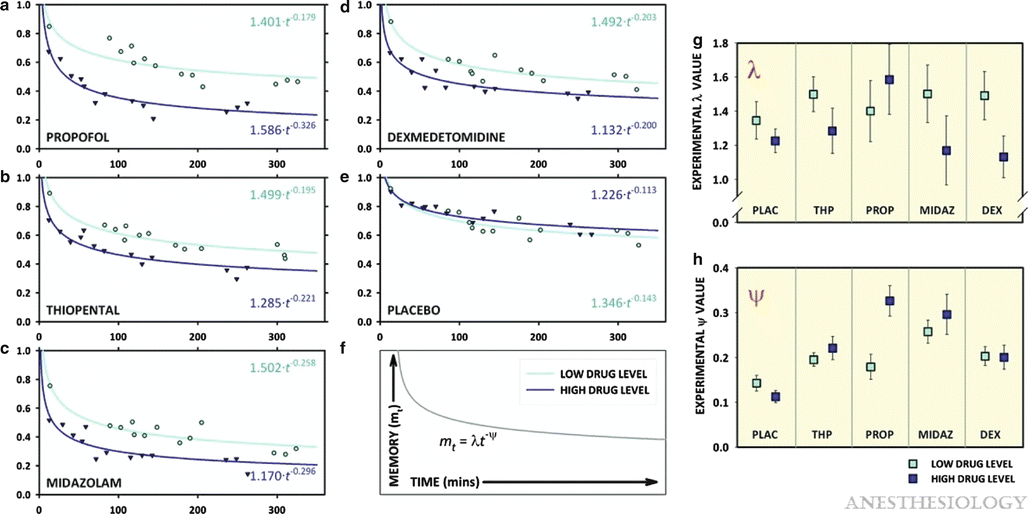
Fig. 10.6
Memory decay curves and estimates for λ (lambda) and ψ (psi) by drug and dose condition. (a–f) The power decay curves for each of the drug–dose combinations. The abscissa represents the duration from the final encoding exposure to the subsequent recognition exposure (the recognition interval). For each curve, recognition data were corrected for false positives, clustered into 13 time points, and then fit to the equation mt = λ (lambda) t − ψ (psi) using Marquardt–Levenberg nonlinear regression. The specific equations describing each curve are shown in the upper and lower right corners of panels a–f. (g, h) The values for λ (lambda) and ψ (psi) are shown, with error bars representing the SEM for the estimate derived from the nonlinear regression. However, statistical comparison of the decay functions was performed using a more robust nonlinear mixed effects model. For λ (lambda), independent effects were established for drug (P < 0.0001) and level (P < 0.0001) and significant fixed effects seen for the midazolam to level interaction (P = 0.014), the dexmedetomidine to level interaction (P = 0.008), and the propofol to level interaction (P = 0.008). For ψ (psi), independent effects were established for drug (P = 0.027) and drug–level interaction (P < 0.0001), whereas fixed effects were established for midazolam (P = 0.008) and propofol to level interaction (P < 0.0001). DEX dexmedetomidine, MIDAZ midazolam, PLAC placebo, PROP propofol, THP thiopental (Reprinted with permission from Pryor KO, Reinsel RA, Mehta M, Li Y, Wixted JT, Veselis RA. Visual p2–n2 complex and arousal at the time of encoding predict the time domain characteristics of amnesia for multiple intravenous anesthetic drugs in humans. Anesthesiology. 2010 Aug;113(2):313-26)
As mentioned, consolidation represents dozens of physiologic processes, each coming online at a given time after learning. Let us say that this time is T critical for a given process, P critical. If propofol exerts its amnesic effects because it impairs P critical, then it will also block any memories that have not yet reached the P critical stage, in other words those memories “in the pipeline.” Memories are being continuously formed, and at any given time, a certain number of memories are in the pipeline before P critical. P critical is continuously processing memories, including the moment propofol is administered at time zero, t = 0. It stands to reason that when propofol is given, then any memories in the pipeline at t = 0 will be lost. In other words, we can determine T critical by measuring the time at which memories are lost before propofol was administered. This type of memory loss is called retrograde amnesia and is commonly observed in cases of severe head trauma (which might be considered in a sense as a “computer crash”). It is also observed in medically induced therapy that “reboots the computer,” namely, electroconvulsive therapy [107, 108]. These major insults to memory processing can be thought of as stopping all consolidation processes at once, and the length of retrograde amnesia gives some indication of how long it takes for a new memory to become an old memory, resistant to various insults2. The key observation of drug-induced amnesia is that despite very careful observations, no significant retrograde amnesia has ever been measured [109]. Thus, the P critical affected by amnesic drugs must be a consolidation process that occurs within minutes after learning a new memory. A likely candidate process is one that is based on electrophysiologic processing of incoming information [98, 110, 111].
As discussed previously in the section “Clinical Practice”, in reality propofol is rarely administered to produce amnesia; rather it is given at higher doses to produce sedation. Even then, no evidence of retrograde amnesia is present.
Benzodiazepines
The classic amnesic drug is the benzodiazepine originally marketed to manage anxiety, namely, diazepam. For many years, benzodiazepines were known to have side effects that included memory impairment. As these drugs were originally used to manage anxiety, this side effect was considered annoying, and great efforts were expended to eliminate this quality, which continue to this day [112, 113]. Initially, memory impairment was considered to be primarily related to the sedative effects of these drugs, and nonsedating benzodiazepines were sought [114]. Eventually these drugs were introduced into situations of extreme anxiety, such as when invasive procedures were necessary, treatment in the critical care unit, or surgery. In the operating room, benzodiazepines were used as adjuncts to other anesthetics as benzodiazepines themselves could not induce an anesthetic state even with large doses. Clinical practice evolved from using these drugs to induce anesthesia, where significant hangover effects occurred due to the relatively long half-life of even “short-acting” benzodiazepines, to using lower doses to induce sedation and anxiolysis [115]. When used in this fashion, a curious observation was noted. Patients receiving diazepam, and subsequently midazolam, would be calm and sedated, but would ask the same question over and over. It became evident that the patient would have no recollection of the answer provided and would then ask the same question repeatedly. This anecdotal observation started further detailed inquiry into the exact nature of the memory effects of sedative drugs used in the hospital setting. As has happened with so many drugs, an annoying side effect became the main therapeutic indication for that drug. These were ideal agents, more so in adults than pediatric patients, as when these drugs were given almost no recollection of events during sedation occurred. The distinction between sedation and actual amnesia was born rather dramatically. For example, after a few milligrams of midazolam, the patient could undergo a “rough” bronchoscopy with coughing, yet when they came back for their next procedure, they would comment how lovely their previous experience had been. Careful comparison of midazolam with propofol revealed essentially identical actions on memory processes [54, 56].
Midazolam is frequently used in clinical practice, and a key question is what to expect in terms of amnesia. How much drug to give and how “dense” will the amnesia be? Though the answer is deterministic, in practice response is much more variable. The degree of amnesia is related to the brain concentration of drug, and this can be determined fairly precisely using pharmacokinetic and pharmacodynamic relationships (as done in our volunteer studies). However, all these measures are after the fact. As noted elsewhere, the determination of amnesia is a retrospective measure. We do not know how much memory loss there was until after the fact when we have tested for recognition of events during drug effect. Thus, it is very difficult to determine before a drug is given exactly how much is needed to produce a satisfactory amnesic effect for a given stimulus/procedure in a particular patient. The best that can be done is to rely on previous experience, both personal and published literature. The most reliable effects will be obtained when a predictable blood concentration can be obtained. This is most likely when midazolam is administered intravenously or, a close second, intranasally. Oral administration is less reliable, and if dense amnesia is desired, then likely a relative overdose will be needed that may produce substantial sedation and hangover effects. A few studies have been done in children where memory has been closely assessed in relation to dosing [116–119]. The dose of midazolam used in three of these studies was 0.2 mg/kg intranasally (buccally), and in the study by Kain et al., more than twice the dose, 0.5 mg/kg, was given orally. This study also assessed memory at multiple time points (5, 10, and 20 min). Memory impairment was quantitated as a percent of items recalled spontaneously or recognized (which is always more than recalled) versus a control (placebo or baseline) condition. With intranasal/buccal administration, approximately 25–30 % of items were recalled (recognition of about twice the number of stimuli occurred when tested), whereas 0 % were recalled (40 % recognized) for oral administration in the Kain et al. study. The peak effect for amnesia with oral administration was at 20 min (but further time points were not tested). Importantly, it should also be noted that items used in all these studies were non-salient (e.g., pictures). Thus, in summary, the published literature showed that with these doses and routes of administration for midazolam, amnesia was significant, but not necessarily “dense.” It is likely that recall for emotive or painful stimuli would be greater. If dense amnesia is required with rapid recovery, propofol is a much easier drug to titrate, as the sedative effect can be used as a proxy for a likely amnesic effect in real time.
Dexmedetomidine
As described in the introduction, the ideal sedative agent would produce a state similar to natural sleep without any drug “hangover,” be easily titratable, and be safe enough (i.e., minimal respiratory compromise), so that non-anesthesia personnel could administer it. Dexmedetomidine comes close to fulfilling these highly desirable goals [120] (however, see the study by Oto et al. [121] that demonstrated abnormal sleep patterns with dexmedetomidine sedation—but this was a study in ICU patients and may not be relevant to other patient populations). Of all the sedative agents available, dexmedetomidine comes closest to targeting natural sleep pathways, with primary actions upstream from modulation of GABAergic receptors in the sleep pathways [14]. However, as opposed to the GABAergic agents propofol and the benzodiazepines, there seems to be little effect on memory per se, other than that produced by sedation [54, 85, 122]. This is likely due to the fact that dexmedetomidine does not affect GABA receptors directly, thus having little effect on these receptors in the medial temporal lobe. The phrase “there seems” is used as very little investigation has been undertaken to determine if dexmedetomidine is an amnesic agent, as defined previously in this chapter. What little evidence there is supports dexmedetomidine as a sedative agent with little amnesic effect. If the “forgetting” characteristics of dexmedetomidine are compared side by side with midazolam, propofol, and thiopental (a short-acting barbiturate that has sedative actions on memory, no longer available for clinical use), it is found that these resemble thiopental more than the amnesic “forgetting” profile of midazolam or propofol [54]. Similarly, the electrophysiologic signature of the effect of dexmedetomidine on memory processes is again different than the amnesic drugs midazolam and propofol [85]. Thus, mechanistically, actions of dexmedetomidine on memory processes are different than typical amnesic drugs. Whether this translates into clinically appreciable differences is still an open question, but retrospective analysis of a large post-op Quality Assurance database revealed cases of recall of intraoperative events associated with dexmedetomidine, but not propofol (in this case, during cardiac surgery), providing indirect evidence of the lack of amnesic actions of dexmedetomidine [122].
Due to its upstream action at the locus ceruleus, in turn modulating GABA receptors in the VLPO, the quality of sedation is different than other GABAergic agents, notably midazolam and pentobarbital, which directly target GABA receptors [14, 17]. Similar to sleep, there can be rapid transitions from deep sedation to wakefulness, but these may not be entirely predictable. Also, during induction of sedation, which requires administration of a bolus of drug over about a 10-min period, the absence of stimulation is important in ensuring a smooth induction of sedation. As with its parent compound clonidine, which significantly potentiates but cannot act alone as an anesthetic drug, dexmedetomidine may find its most useful niche as a major component of a multidrug regimen. For example, the analgesic effect of ketamine may help prevent movement during procedures, and the side effect of tachycardia with ketamine may counteract the propensity for bradycardia from dexmedetomidine [123].
Ketamine
As with all drugs discussed in this chapter, what we know about ketamine’s effects on cognition is derived from adults. Ketamine has a long history of use in pediatrics, with its use falling dramatically with the arrival of newer agents. The pendulum is swinging back with ketamine being used as an adjunct to other sedatives. Ketamine’s side effects are often complementary with the side effects of other drugs. For instance, the amnesic properties of ketamine may be beneficial when used with dexmedetomidine, which seems to have poor amnesic effects beyond its sedative properties. Additionally, ketamine has potentially beneficial analgesic and antiinflammatory properties when given in low doses [124–126].
Ketamine has substantial effects on cognition, and these have been examined from the perspective of psychiatry and drug addiction fields. As a consequence, the cognitive effects of ketamine are much better understood than, for instance, dexmedetomidine or etomidate. Ketamine has been used to model the cognitive changes occurring in schizophrenia, such as hallucinations, delusions, idiosyncratic and illogical thinking, poverty of speech and thought, agitation, disturbances of emotion and affect, withdrawal, decreased motivation, and dissociation [127, 128]. It should be noted that many of these “disturbing” symptoms are uncommon in the clinical setting. These disturbing psychologic side effects are more likely to be seen when ketamine, which is one of the big three “club” drugs, is used in unmonitored settings (the “K-hole”) [129–131].
There is good evidence that ketamine is a true amnesic drug as opposed to simply being a sedative agent. It produces substantial memory impairment, at doses 0.4–0.8 mg/kg that produce little sedation [132]. Some studies have administered ketamine as a continuous infusion that maintained fairly constant serum concentrations over the time period of memory encoding, similar to our studies measuring the memory effects of propofol [133, 134]. Results from these studies can be interpreted using the same schema for memory as used with propofol, as presented earlier. Doses of ketamine that produced serum concentrations of 129 ng/mL produced similar degrees of sedation as did propofol in our studies, as measured by increases in reaction times. In other words, these doses of ketamine were equi-sedative to the amnesic doses of propofol we have studied. In these studies, ketamine produced a similar characteristic of forgetting of memories encoded in the presence of drug over time as previously demonstrated for propofol.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


