1. Rule out treatable causes of agitation
(a) Hypoxia and hypercarbia
(b) Cerebral hypoperfusion
(c) Bladder distention
(d) Surgical lesion—necrotic bowel or compartment syndrome
2. Perform a presedation evaluation of the patient. This evaluation is similar to that performed prior to any surgical procedure performed in the operating room
3. Identify the etiology of the pain or agitation to guide the appropriate selection of the agent or agents as well as the need to provide sedation/anxiolysis/amnesia, analgesia, or both
4. Monitor patient according to the standards outlined by the American Academy of Pediatrics for procedural sedation and analgesia
5. Titrate the initial bolus dose of the medication and subsequent infusion rates based on the patient’s clinical response with the use of formalized sedation/pain scales
6. Observe for adverse physiologic effects including the development of physical tolerance which necessitates increasing the dose of the agent used or switching to another agent that acts through a different receptor system
The basic components of the presedation assessment are outlined in Table 16.2. This assessment includes the performance of a focused history and physical examination. The history should focus on the child’s current state of health as it relates to the reason for the procedure, the past medical history to identify significant comorbidities, as well as acute events that led to the PICU admission. Since the primary risks associated with sedation include adverse respiratory events (apnea, hypoxemia, hypercarbia, and upper airway obstruction) or cardiovascular events (hypotension, bradycardia, arrhythmias), the focus of the presedation evaluation and physical examination is placed on these systems or areas. The American Society of Anesthesiologists (ASA) emphasizes that the evaluation of the airway “should be conducted, whenever feasible, prior to the initiation of anesthetic care and airway management in all patients”[2]. The purpose of this examination is to detect physical characteristics that may indicate the presence of a difficult airway. (Refer to Chap. 7.) Although many patients may already have an endotracheal tube in place, the assessment of the upper airway should still be performed in the event that the endotracheal tube becomes dislodged at some time, such as during positioning for the procedure or patient transport. The upper airway assessment includes obtaining a history of signs and symptoms that indicate potential airway problems (noisy breathing, stridor, snoring, or obstructive sleep apnea). This is followed by an examination of the head and neck designed to identify the patient in whom airway management or endotracheal intubation may be difficult. An assessment is made of neck mobility (flexion and extension), mouth opening, the size of the oral cavity and tongue (presence of macroglossia), the presence of micrognathia, and the thyromental distance (distance from the thyroid cartilage to the tip of the mandible). In general, mouth opening should be greater than the width of two fingerbreadths, while the thyromental distance should be at least three. In both instances, the patient’s own fingers can be used as the measuring instrument. In the adult population, an objective measure of the potential for difficult intubation is the Mallampati grading system (I through IV) (see Fig. 16.1) [3]. This system evaluates the ability to visualize the tonsillar pillars and the uvula when the patient opens their mouth. If the Mallampati grade is III or IV (tonsillar pillars and base of the uvula cannot be visualized with mouth opening and tongue protrusion), the trachea may be difficult to intubate and bag-mask ventilation may be problematic.
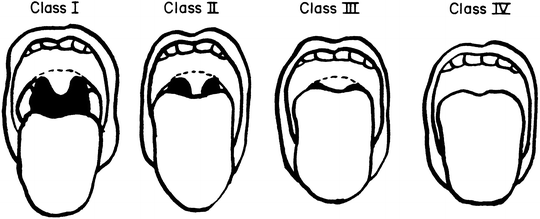
Table 16.2
The pre-procedure or presedation assessment
Patient’s name, age, weight, and gender |
Past medical history: |
Acute medical or surgical problems |
Comorbid medical conditions: |
Previous sedation or anesthetic history including problems |
Allergies |
Current medications |
Family history of anesthetic complications |
Dietary history (nil per os status) |
Pregnancy history |
Physical examination: |
Baseline vital signs including room air oxygen saturation if feasible |
Airway examination with Mallampati grading system |
Focused cardiac and respiratory examination |
Current vascular access and infusion (to select site for medication administration) |
Laboratory evaluation as appropriate |
Summary: |
American Society of Anesthesiologists status (ASA I–V) |
Sedation and recovery plan |
Risks discussed and informed consent obtained from patients |
Perform formal, hospital-based time-out prior to the procedure |
Fig. 16.1
Mallampati classification of pharyngeal structures. Reprinted with permission from Samsoon GL, Young JR. Difficult tracheal intubation: a retrospective study. Anaesthesia. May 1987;42(5):487–490
In the pediatric ICU patient, a thorough review of the patient’s current medical regimen (both sedation/analgesic and others) is performed as well as a determination of the patient’s current vascular access and which site will be used for the administration of the sedative/analgesic agents. Upon completion of the history and physical examination, an ASA (American Association of Anesthesiologist) classification may be assigned (Table 16.3) [4] to help guide the choice of sedation: Adverse events have been shown to have a greater incidence in patients with a higher ASA classification (III or IV versus I or II) [5]. During this evaluation, the child’s previous experiences with procedural sedation are explored, identifying both their efficacy and the child’s and parent’s impressions of the experience. (Refer to Chap. 4.) A final component of the presedation assessment is to minimize the risk of aspiration by adhering to the nil per os (NPO) status in order to limit gastric volume. In the PICU patient, factors other than NPO may affect gastric emptying and thereby both gastric volume and pH. The American Society of Anesthesiologists’ recommendations for per os (NPO) are 2–3 h for clear liquids (including breast milk) and 4–8 h for infant formula [4]. The need to adhere to strict NPO guidelines for procedural sedation have been challenged, particularly by those working in acute care environments such as emergency rooms where procedures may need to be performed more urgently [6, 7]. While published reports from these environments have failed to show an effect of pre-procedure fasting on the incidence of adverse outcomes, these studies have been underpowered. If a patient is considered to be at significant risk of aspiration, consideration should be given to recruit anesthesia to perform a rapid sequence induction and endotracheal intubation. Following a careful assessment and prior to the start of the procedure, a formal “time out” should be performed and documented. This “time out” should include two patient identifiers (name and medical record number), validation that consents have been obtained and signed, a review of the procedure to be performed, and agreement on the site and laterality of the procedure.
Table 16.3
ASA classification
ASA I | No associated comorbid disease |
ASA II | Mild associated comorbid disease, mild asthma |
ASA III | Severe associated comorbid disease, sickle cell anemia, obstructive sleep apnea, severe asthma |
ASA IV | Comorbid disease that is a constant threat to life, dilated cardiomyopathy, septic shock |
ASA V | Moribund patient that is not expected to survive 24 h |
E | Modifier added for emergency procedure or surgery |
Patients should be monitored in accordance with the guidelines set forth by the American Academy of Pediatrics (AAP) and/or the ASA both during and following the procedure [4, 8]. (Refer to Chap. 2.) As the PICU provides the optimal environment for the monitoring of a patient’s physiologic functions, the monitoring should be continued when PICU patients are sedated outside of the PICU. Formal monitoring should include continuous pulse oximetry and heart rate (via the pulse oximeter or electrocardiography) as well as measurement of respiratory rate and blood pressure at 5 min intervals [4, 9, 10]. In 2011, the ASA amended the Standards for Basic Anesthesia Monitoring (first published in 1986) to specify that during moderate and deep sedation, ventilation should be followed by clinical observation and capnography [11]. Exceptions to capnography would be situations whereby patient, procedure, or equipment precludes or invalidates the monitoring. Adherence to these monitoring guidelines is mandatory as lack of appropriate monitoring has been shown to be a key failure in the analysis of adverse outcomes during procedural sedation [12]. Monitoring of ventilation via end-tidal carbon dioxide (ETCO2) should also be considered under conditions where access to the patient is limited. ETCO2 monitoring has been shown to detect hypercarbia in the absence of clinically apparent respiratory depression or desaturation if only pulse oximetry is used [13, 14]. ETCO2 provides a tangible means to monitor and adjust ventilation in the intubated child. Ventilatory regulation in order to maintain a targeted PaCO2 can be particularly important for patients with altered intracranial compliance, pulmonary hypertension, or those with compromised or cyanotic heart disease.
Assessing the Depth of Sedation
Some means for the ongoing evaluation of the depth of sedation should be incorporated into the procedural sedation routine. This monitoring allows for an increase or decrease in the doses used, based on the patient’s depth of sedation rather than the hemodynamic parameters. Subjective scoring measures and assessments have been replaced by standardized pain and sedation scoring systems, which are monitored at regular intervals. (Refer to Chap. 5.) These scoring systems should be used both during prolonged sedation as well as brief periods of procedural sedation. The currently used PICU sedation scores evaluate physiologic variables, an objective assessment of the patient’s depth of sedation, or a combination of the two. One commonly used scale, the COMFORT score, combines the scoring of a patient’s response or movement in addition to various physiologic parameters (see Table 16.4) [15]. The COMFORT score assesses alertness, respiratory rate, blood pressure, muscle tone, agitation, movement, heart rate, and facial tension. It has been validated in the pediatric-aged patient and may have utility in the assessment of sedation during mechanical ventilation [15, 16]. Beware that scales based on physiologic parameters can be misleading in an ICU. Furthermore, patients with cardiovascular dysfunction requiring vasoactive medications may not manifest increases in heart rate and blood pressure normally seen with severe agitation or pain. Because of these concerns, Ista et al. have recently proposed the COMFORT-B score (see Table 16.5), a modification of the original COMFORT score, which eliminates physiologic variables and provides new cutoff points for the diagnosis of over- or undersedation [17].
Characteristic | Evaluate | Points |
|---|---|---|
Alertness | Deeply asleep | 1 |
Lightly asleep | 2 | |
Drowsy | 3 | |
Awake and alert | 4 | |
Hyperalert | 5 | |
Agitation | Calm | 1 |
Slightly anxious | 2 | |
Anxious | 3 | |
Very anxious | 4 | |
Panicky | 5 | |
Respiratory response | No coughing | 1 |
Spontaneous respiration with little response to ventilation | 2 | |
Occasional coughing with little resistance to the ventilator | 3 | |
Active breathing against the ventilator | 4 | |
Actively fighting the ventilator and coughing | 5 | |
Physical movements | None | 1 |
Occasional, slight movements | 2 | |
Frequent, slight movements | 3 | |
Vigorous movements of extremities only | 4 | |
Vigorous movements of extremities, torso, and head | 5 | |
Blood pressure (mean) | Below baseline | 1 |
Normal | 2 | |
Infrequent elevations of 15 % or more | 3 | |
Frequent elevations of 15 % or more | 4 | |
Sustained elevation greater than or equal to 15 % | 5 | |
Heart rate | Below baseline | 1 |
Normal | 2 | |
Infrequent elevations of 15 % or more | 3 | |
Frequent elevations of 15 % or more | 4 | |
Sustained elevation greater than or equal to 15 % | 5 | |
Muscle tone | Relaxed/none | 1 |
Reduced muscle tone | 2 | |
Normal muscle tone | 3 | |
Increased tone/flexion—fingers/toes | 4 | |
Extreme rigidity/flexion—fingers/toes | 5 | |
Facial tensionTotal: | Facial muscles relaxed | 1 |
Normal tone | 2 | |
Some tension | 3 | |
Full facial tension | 4 | |
Facial grimacing | 5 |
Table 16.5
The COMFORT-B scale
AlertnessPonits |
Deeply asleep1 |
Lightly asleep2 |
Drowsy3 |
Fully awake and alert4 |
Hyperalert5 |
Calmness/agitation |
Calm1 |
Slightly anxious2 |
Anxious3 |
Very anxious4 |
Panicky5 |
Respiratory response (ventilated children) |
No coughing and no spontaneous respiration1 |
Spontaneous respiration with little or no response to ventilation2 |
Occasional cough or resistance to ventilator3 |
Actively breathes against ventilator or coughs regularly4 |
Fights ventilator, cough, or choking5 |
Cry (non-ventilated children) |
Quiet breathing, no crying1 |
Sobbing or gasping2 |
Moaning3 |
Crying4 |
Screaming5 |
Physical movement |
No movement1 |
Occasional, slight movements2 |
Frequent, slight movements3 |
Vigorous movement limited to extremities4 |
Vigorous movements including torso and head5 |
Muscle tone |
Muscles totally relaxed, no muscle tone1 |
Reduced muscle tone2 |
Normal muscle tone3 |
Increased muscle tone and flexion of fingers and toes4 |
Extreme muscle rigidity and flexion of fingers and toes5 |
Facial tension |
Facial muscle totally relaxed1 |
Facial muscle tone normal; no facial muscle tension evident2 |
Tension evident in some facial muscles3 |
Tension evident throughout facial muscles4 |
Facial muscles contorted and grimacing5 |
Total: |
Other scoring systems such as the sedation–agitation scale (SAS) (see Table 16.6) also eliminate the use of physiologic parameters by visually assessing the level of the patient’s comfort and grades it from 1 (unarousable) to 7 (dangerous agitation such as pulling at the ETT) [18].
Scale | Agitation/sedation | Behavior |
|---|---|---|
7 | Dangerous agitation | Pulls at endotracheal tube, tries to remove catheters, climbs over bedrail, strikes at staff, thrashes side to side |
6 | Very agitated | Does not calm despite frequent verbal reminders of limits, requires physical restraint, bites endotracheal tube |
5 | Agitated | Anxious or mildly agitated, attempts to sit up, calms down to verbal instructions |
4 | Calm and cooperate | Calm, awakens easily, follows commands |
3 | Sedated | Difficult to arouse, awakens to verbal stimuli or gentle shaking but drifts off again, follows simple commands |
2 | Very sedated | Arouses to physical stimuli but does not communicate or follow commands, may move spontaneously |
1 | Unarousable | Minimal or no response to noxious stimuli, does not communicate or follow commands |
The Ramsay scale, a sedation scale used commonly in the adult ICU population, also assigns a value based on the observation of the patient, but also uses a tactile stimulus (a glabellar tap) to distinguish between the deeper levels of sedation [19]. Scoring for the Ramsey scale varies from 1 (awake, anxious, and agitated) to 6 (no response to a glabellar tap). The Hartwig score similarly uses a visual assessment of the patient, but as with the Ramsay scale includes a response to a noxious stimulus, in this case, tracheal suctioning thereby eliminating its use in non-intubated patients [20]. Scales such as the Ramsay and the Hartwig that assess the response to a tactile stimulus require disturbing the patient to differentiate between the deeper levels of sedation. Additionally, scales that evaluate a patient’s response to a stimulus or observe their behavior are not valid during the administration of neuromuscular blocking agents that prevent movement.
Various scales have also been developed for assessing the patient during procedural sedation. Given the potential morbidity associated with patients becoming too deeply sedated as well as concerns of inadequate sedation, tools that provide an accurate assessment of the response to sedative and analgesic agents during procedural sedation are also needed. During light sedation, this may be easily accomplished by assessment of the patient’s ability to appropriately respond to questions. However, with deeper sedation, such assessments become of limited utility. The Observer’s Assessment of Alertness/Sedation (OAAS) scale has been validated in children, but has been shown to have a limited ability to differentiate between the deeper levels of sedation [21]. The Vancouver Sedative Recovery Scale appears to be better able to differentiate deeper levels of sedation, although it is likely too cumbersome to be easily utilized during short procedures [22]. More recently, Malviya et al. developed and validated the University of Michigan Sedation Scale (UMSS) (see Table 16.7) [23]. This scale was developed to be a simple and efficient tool to assess depth of sedation over the entire sedation continuum and one that could easily be applied by various health-care providers. It utilizes a simple scale ranging from 0 (awake and alert) to 4 (unresponsive).
Table 16.7
The University of Michigan Sedation Scale (UMSS)
Scale | Level of sedation |
|---|---|
0 | Awake and alert |
1 | Minimally sedated |
• Tired/sleepy | |
• Appropriate response to verbal conversation and/or sounds | |
2 | Moderately sedated |
• Somnolent/sleeping | |
• Easily aroused with light tactile stimulation or simple verbal commands | |
3 | Deeply sedated |
• Deep sleep | |
• Arousable only with significant physical stimulation | |
4 | Unarousable |
In addition to the classical sedation scales, there may be a role for monitoring technology to assess the depth of sedation through the analysis of the electroencephalogram (EEG). The bispectral index (BIS monitor, Covidien, Boulder, Colorado) uses an algorithm to interrogate the processed EEG pattern and provide a numeric value ranging from 0 (isoelectric) to 100 (awake with eyes open). Its predominant clinical use has been intraoperatively to monitor the effects of general anesthetic and sedative agents and provide a measure of the depth of anesthesia. (Refer to Chap. 6 .) Although still somewhat controversial, it has been suggested that maintenance of a BIS value less than 60–70 correlates with a low probability of intraoperative awareness [24, 25]. The BIS monitor has also been used in settings outside of the operating room including the ICU where assessment of sedation is critical to interventions such as procedural sedation or mechanical ventilation [26–34]. Gill et al. compared BIS values with Ramsay sedation scores in 37 adults who received procedural sedation in the emergency room setting [26]. Although the BIS number correlated with the depth of sedation, there was a wide variability in BIS values at similar sedation scores. The BIS was most effective at differentiating moderate-to-deep sedation from general anesthesia. Brown-McDermott et al. compared BIS values with UMSS scores during procedural sedation administration in 86 children less than 12 years of age [27]. Although there was a good correlation between the BIS value and the sedation score even in patients less than 6 months of age, a wide variability in the range of BIS values for each level of sedation was again noted. Additionally, the BIS monitor was ineffective at determining the depth of sedation when specific agents were used, including ketamine or a combination of oral chloral hydrate, hydroxyzine, and meperidine. The latter is not surprising as the algorithm for the BIS monitor was developed during the use of general anesthetic agents such as propofol or the inhalational anesthetic agents that act through the GABA (gamma-aminobutyric acid) system.
Despite these shortcomings, the BIS monitor may be able to effectively identify patients who are becoming too deeply sedated and may therefore be at risk for adverse respiratory events. Motas et al. demonstrated that the depth of sedation as judged by the BIS monitor was predictive of adverse airway events during the administration of procedural sedation (either propofol, midazolam, or pentobarbital) by non-anesthesiologists [28]. The number of episodes of oxygen desaturation and airway events respectively increased from 1 and 0 of 20 patients when the BIS number was 71–90 to 2 and 3 of 17 patients when the BIS number was 61–70 and to 4 and 4 of 24 patients when the BIS number was less than 60.
BIS monitoring has also been evaluated as a means of evaluating the depth of sedation during prolonged mechanical ventilation. Although the results have been somewhat mixed, the majority of reports have demonstrated a clinically acceptable correlation between the BIS monitor and commonly used ICU sedation scores including the Ramsay or the COMFORT score [29–33]. As with its use during procedural, it has generally been shown that the BIS number correlates with the depth of sedation as assessed with the clinical sedation score, although there is significant variability in the BIS number at each level of sedation. Part of this variation may be related to interference from electromyographic (EMG) artifact from facial musculature, which may falsely elevate the BIS number [34, 35]. The more recent versions of the BIS probe incorporates a sensor that may be able to effectively eliminate EMG interference from the BIS algorithm and thereby address this issue. More importantly, the BIS algorithm was developed for use with propofol or the potent inhalational anesthetic agents that work through the GABA system. Therefore, the BIS monitor has not been shown to be an accurate means of judging the depth of anesthesia with the administration of etomidate or agents that act through the N-methyl-d-aspartate (NMDA) system including xenon or nitrous oxide [36–38].
Despite these issues, some form of depth of sedation monitoring may be clinically helpful in situations that preclude the use of conventional ICU scoring systems such as patients receiving neuromuscular blocking agents and/or medications that may alter heart rate and blood pressure responses and thereby negate the utility of clinical sedation scoring systems [39–42]. Additionally, technology such as the BIS monitor provides a continuous numeric readout using a simple 0–100 scale that is immediately available at the bedside as opposed to sedation scoring systems that provide only an intermittent assessment and require time to assess and add various parameters.
Basic Principles
Identification of the cause of the distress can be used to guide the selection of the most appropriate agent(s) for sedation and analgesia in the PICU. Tissue injury, an acute inflammatory process, or performance of a painful invasive procedure is generally best treated by an agent with analgesic properties, while emotional distress and anxiety may be more appropriately treated with agents that possess sedative, anxiolytic, and amnestic properties. Another variable is the duration of time during which sedation is required. Sedation may be required for less than 5 min for an invasive procedure, 1–2 h during an MRI scan, or days to weeks in a patient requiring prolonged mechanical ventilation or extracorporeal support. Short-acting drugs that provide rapid recovery may be chosen for brief procedures, while intermittent dosing of longer-acting agents or continuous infusions of short-acting agents can be used for prolonged procedures. There may be associated comorbid diseases that alter the end-organ effects of these agents, their adverse profile, and metabolism or elimination.
To date, there is limited evidence-based medicine from which to develop guidelines for the use of sedative and analgesic agents in the PICU setting. When compared with the adult population, there remains a paucity of data regarding the pharmacokinetics and pharmacodynamic properties of analgesic and sedative drugs in critically ill infants and children [43–45]. When available, these pharmacokinetic studies are generally performed in healthy adult volunteers with the attempted extrapolation of these results to critically ill infants and children. The comorbidities present in the PICU may affect the volume of distribution and elimination half-life, thereby further altering the pharmacokinetics of these agents. Additional variabilities in the PICU setting are likely to result from drug–drug interactions, end-organ (hepatic, renal) failure or dysfunction, malnutrition, low plasma proteins with altered drug binding, alterations in uptake of the medication if non-intravenous routes are used, and alterations in drug distribution.
Pharmacogenetic factors may also affect responses to medications including the response to acute illness and the metabolism and elimination of various medications [46, 47]. These genetic variations may also alter drug receptors and the patient’s response to various sedative and analgesic agents [48, 49]. As there are currently limited means of identifying the impact of these many factors on the pharmacokinetics and pharmacodynamics of the medications that we use for sedation and analgesia in the PICU setting, it is imperative that the effects of these agents are continuously evaluated and the dose titrated based on the patient’s response. An example of such variability, likely a multifactorial phenomenon in the PICU population, is demonstrated by an evaluation of fentanyl infusion requirements to provide sedation during mechanical ventilation in neonates and infants [50]. To achieve a similar level of sedation, the fentanyl infusion requirements varied from 0.47 up to 10.3 μ(mu)g/kg/h. Therefore, it is not feasible to approach the provision of sedation and analgesia in the PICU patient using a “cookbook” with specific guidelines concerning the medications to be used and their doses. Sedative and analgesic agents cannot be dosed on a per kilogram basis; rather, the infusion rate should be titrated up and down to achieve the desired level of sedation. The dosing recommendations provided in this chapter for the specific medications discussed are meant only as guidelines for starting doses. The actual dose should be titrated up or down to achieve the desired level of sedation or analgesia while attempting to avoid adverse effects [51].
More recently, the variability and the potential for adverse effects including prolonged sedation have led to the suggestion of using “drug holidays” on a daily basis. This process involves discontinuing the administration of sedative and analgesic agents until the patient awakens and then restarting the infusions at 50–75 % of the previous rate. In a prospective trial in 128 adults requiring mechanical ventilation, the median duration of mechanical ventilation was 4.9 days in the intervention group and 7.3 days in the control group (p = 0.004) [52]. The median length of stay in the intensive care unit was 6.4 days as compared with 9.9 days, respectively (p = 0.02). Six of the patients in the intervention group (9 %) underwent diagnostic testing to assess changes in mental status as compared with 16 or 27 % of the patients in the control group (p = 0.02). Complications (including removal of the endotracheal tube by the patient) occurred in three of the patients in the intervention group and four of the patients in the control group (p = NS). The demonstration of improvements in outcome with such interventions has led to the widespread adoption of such practices in the adult population. Such therapy has been championed by the ICU physicians at Vanderbilt University with the development of the ABCDE bundle. This involves a group of ICU measures including spontaneous awakening and breathing coordination, attention to the choice of sedation, delirium monitoring, and early mobility and exercise. Additional work has demonstrated the potential impact of focusing on titration of ventilatory assistance to achieve synchrony with the patient rather than pushing sedation to eliminate spontaneous ventilation [53].
Despite the potential difficulties and risks of sedation and analgesia in the PICU patient, there may be significant benefits. Aside from humanitarian concerns, clinical trials have reported decreases in morbidity and mortality based on the analgesic regimen following cardiovascular surgery for congenital heart disease in neonates and infants [54, 55]. These effects are postulated to be the result of blunting of the endogenous physiologic stress response thereby decreasing release of endogenous catecholamines and adrenal cortical hormones. This physiologic stress response, when excessive, may have deleterious end-organ effects. Aside from its effects on the physiologic stress response, analgesia and sedation may provide benefits in other clinical scenarios in the PICU patient. Techniques for cardiac and respiratory support such as permissive hypercapnia, reverse I:E ratio ventilation, high-frequency ventilation, and extracorporeal support may be feasible only with effective control of the patient’s agitation. Analgesia and sedation may also provide therapeutic benefits in the treatment of intracranial hypertension or to modulate pulmonary vascular resistance (PVR) in patients at risk for pulmonary hypertension. The effective use of sedation and analgesia may limit the need for neuromuscular blocking agents and their associated adverse effects [56].
Choice of Agent and Route of Delivery
The three primary decision points for sedation and analgesia in the PICU include the agent, its route of administration, and its mode of administration. The remainder of this chapter will focus on a brief discussion of each agent, its pharmacology, clinical applications, and adverse effect profile. Although the intravenous route is chosen in most clinical scenarios, alternative routes may be required in specific clinical scenarios or patient populations. Furthermore, there is expanding knowledge and interest regarding the use of alternative routes, especially inhalational anesthesia or subcutaneous administration in the PICU setting. These routes may be chosen as the primary route or used as an alternative when drug incompatibilities or limited venous access precludes intravenous administration. However, the availability of alternative routes is agent specific. Chloral hydrate must be administered by the oral or rectal route, isoflurane requires inhalation administration, while propofol is administered only via the intravenous route. Midazolam and ketamine provide the greatest options for route of administration with reports of intravenous, intramuscular, subcutaneous, oral, nasal, and sublingual administration. The third decision point is the mode of administration. Options include continuous administration, intermittent dosing, or patient-controlled techniques. When there is a prolonged need for sedation such as during mechanical ventilation, agents with long elimination half-times may be used by intermittent, bolus administration, while short-acting agents are generally best administered by a continuous infusion to maintain a steady-state serum concentration and thereby provide a stable level of sedation.
Inhalational Anesthetic Agents
Despite their introduction into clinical practice more than 150 years ago, the potent inhalational anesthetic agents remain one of the integral components for the provision of general anesthesia. They are used on a daily basis during the perioperative period to provide amnesia and analgesia during major and minor surgical procedures. Based on their chemical structure, these agents can be divided into alkanes such as halothane or substituted ethers. Although it was the mainstay of pediatric anesthesia for decades, halothane is no longer commonly used and will not be discussed in great detail. The agents in use include the methyl, ethyl ethers (isoflurane, desflurane) and the isopropyl ether, sevoflurane. The characteristics of these agents that may make them useful agents for ICU sedation include a rapid onset, rapid awakening upon discontinuation, and the ability to rapidly control the depth of sedation. As these agents are volatile substances, they are vaporized and administered by the inhalation route. The potent inhalational anesthetic agents also provide specific therapeutic end-organ effects including bronchodilation, myocardial preconditioning, and cerebral protection. Although experience with use of the potent inhalational anesthetic agents for ICU sedation in the United States is limited, certain centers in Europe and the United Kingdom have reported favorable experiences with these agents in adult ICU patients [57]. For a full review of the end-organ effects of these agents, their applications in the pediatric ICU, and techniques for their delivery, the reader is referred to the article by Tobias [58].
Although there are reports concerning the administration of all of the potent inhalational anesthetic agents as either therapeutic agents or for sedation in the ICU setting [59, 60], the majority of reports have used isoflurane. Despite the fact that these agents are all considered in the category of the potent inhalational anesthetic agents, their physiologic effects are distinctly different. Various adverse physiologic effects have been reported with halothane including a negative inotropic and chronotropic effect on myocardial function, the potential for a pro-arrhythmogenic effect especially in the setting of increased catecholamines or when used in conjunction with other medications (e.g., aminophylline), and the potential for the development of hepatitis related to an immunologic reaction directed against the oxidative metabolite, trifluoroacetic acid (TFA) [61, 62]. Hepatitis may occur with any of the inhalational anesthetic agents except for sevoflurane as its metabolic pathway does not result in the production of TFA. The incidence of hepatitis is extremely low with isoflurane compared to halothane due to its limited metabolism of 0.1–0.2 % compared with that of halothane (15–20 %). Given the potential for adverse effects on myocardial function and its association with perioperative cardiac arrests in infants and children, halothane is no longer in use in the United States [63, 64].
Metabolism of the inhalational anesthetic agents can also result in the release of inorganic fluoride and the potential for nephrotoxicity. This was primarily a concern with enflurane, which is no longer used in clinical practice. Although only 2 % of enflurane undergoes metabolic degradation, given its high fluoride content, serum fluoride concentrations increase with prolonged administration. Plasma fluoride concentrations in excess of 50 μ(mu)mol/L can have deleterious effects on renal function with a decreased glomerular filtration rate and renal tubular resistance to vasopressin with nephrogenic diabetes insipidus. Three to five percent of sevoflurane also undergoes metabolism, and like enflurane, sevoflurane is highly substituted with fluoride. Therefore, its prolonged administration can also result in elevated serum fluoride concentrations. Given limited experience with the prolonged administration of sevoflurane, there are no definite data on which to base decisions regarding the duration of administration.
Desflurane is the newest of the inhalational anesthetic agents. Its beneficial properties include low blood to gas and blood to fat solubility coefficients, thereby resulting in a rapid onset and rapid awakening upon its discontinuation. When compared with propofol for postoperative sedation of adults requiring mechanical ventilation, the emergence time was shorter and more predictable and there was a more rapid mental recovery with desflurane with no difference in the incidence of adverse effects [59]. Medication costs were lower with desflurane than with propofol (€95 Euros for desflurane versus €171 Euros for propofol per 24 h) with additional costs of soda lime (€5 Euros) being comparable to the costs of infusion tubing for propofol (€2.5 Euros). Adverse effects with desflurane include hypotension primarily from peripheral vasodilation, rebound tachycardia from stimulation of the sympathetic nervous system, direct irritant effects on the airway thereby making it less than optimal in patients with airway hyperreactivity, and rare reports of carbon monoxide formation due to desflurane’s interaction with desiccated soda lime [65, 66].
Additional concerns with all of the inhalational anesthetic agents include their potential as a trigger agent for malignant hyperthermia, cost issues, effects on intracranial pressure (ICP), and alterations of the metabolism of other medications. As nonspecific vasodilators, all of the inhalational anesthetic agents cause cerebral vasodilation resulting in an increase in ICP in patients with compromised intracranial compliance. The effects on the cerebral vasculature and ICP can be blunted by modest hyperventilation [67, 68]. The inhalational anesthetic agents may alter hepatic blood flow and affect the metabolism of other medications including lidocaine and other local anesthetic agents, ß(beta)-adrenergic antagonists, and benzodiazepines [69].
There remains limited experience with the use of the potent inhalational anesthetic agents for sedation in the PICU setting. Arnold et al. reported their experience with isoflurane for sedation in ten pediatric patients, ranging in age from 3 weeks to 19 years, who required endotracheal intubation and mechanical ventilation for various reasons [70]. Sedation was initiated with isoflurane at an inspired concentration of 0.5 % and adjusted in 0.2 % increments as needed. The duration of administration varied from 29 to 769 h (245 ± 225 h). There was adequate sedation 75 % of the time, excessive sedation 4 % of the time, and inadequate sedation 21 % of the time. In the five patients who received isoflurane for at least 96 MAC (minimum alveolar concentration)-hours, there were no differences in blood urea nitrogen, serum creatinine, osmolality, bilirubin, and alanine aminotransferase between time 0 and 96 h. The duration of isoflurane administration correlated directly with the plasma fluoride concentration. The peak serum fluoride concentration averaged 11.0 ± 6.4 μ(mu)mol/L with a maximum value of 26.1 μ(mu)mol/L after 441 MAC-hours of isoflurane. One patient developed hemodynamic instability, which responded to fluid administration. When it was discontinued, five of the patients, who had received greater than 70 MAC-hours of isoflurane, manifested signs and symptoms of withdrawal including agitation and non-purposeful movements.
Despite the potential advantages of using these agents in the ICU setting, logistic problems regarding delivery continue to impede their widespread introduction into the ICU setting. The exhaled gas from the ventilator or anesthesia machine must be collected or scavenged and vented out of the ICU environment. Additional equipment required for the use of these agents in the ICU includes a vaporizer, which converts the liquid phase of the agent to a gas for delivery, and monitors to measure the end-tidal (exhaled) concentration of the drug. As a full discussion of these issues is beyond the scope of this chapter, the reader is referred to the article by Tobias [58] for a full discussion of delivery options and other considerations regarding the administration of the potent inhalational anesthetic agents in the ICU setting. Given these problems, novel means of delivering these agents are needed.
The Anesthetic Conserving Device or “AnaConDa®” (ACD, Hudson RCI, Upplands Väsby, Sweden) is a modified heat–moisture exchanger that may allow a simplified means of administering the inhalational anesthetic agents in the ICU setting. This device is not currently available in the United States. The device is placed between the Y-piece of the ventilator circuit and the endotracheal tube (ETT). There is also a port at the end of the device just proximal to its attachment to the ETT that allows gas sampling and monitoring of the agent concentration. The desired inspired concentration is titrated by adjusting the infusion rate on the syringe pump based on the manufacturer’s recommendations. Exhaled isoflurane is adsorbed to the lipophilic carbon particle filter in the device and redelivered to the patient thereby limiting environmental pollution. Preliminary work has evaluated the ACD in the ICU setting in 40 adult patients requiring sedation for more than 12 h [71]. The patients were randomized to sedation with isoflurane administered with the ACD or a continuous infusion of midazolam. The inspired isoflurane concentration was started at 0.5 % (infusion rate on the syringe pump of 1–3.5 mL/h according to the manufacturer’s recommendations) while midazolam was infused at 0.02–0.05 mg/kg/h. The infusion rates were adjusted as needed and opioids administered for analgesia. The percentage of time within the desired level of sedation was similar between the two groups (54 % with isoflurane and 59 % with midazolam) with no difference in opioid requirements or the need for bolus doses of sedative agents. The time to extubation (10 ± 5 versus 252 ± 271 min) and the time to follow verbal commands (10 ± 8 versus 110 ± 132 min) were shorter with isoflurane than with midazolam. Isoflurane-sedated patients had normal urine volumes and creatinine clearances. The inorganic fluoride concentration was greater than 50 μ(mu)mol/L in three patients with a maximum value of 64 μ(mu)mol/L during isoflurane sedation. Anecdotal experience with the device has also been reported in three pediatric patients who required sedation during mechanical ventilation or in the treatment of status epilepticus [72]. Effective sedation was achieved with isoflurane concentrations of 0.3–0.4 %, while 0.9 % was required to control status epilepticus. In 2 patients, who weighed 30 and 40 kg, respectively, the ACD was placed distal to the Y-piece as described in the adult population. In the third patient, who weighed 20 kg, when the ACD was placed in this position, the patient became tachypneic with an increase in his respiratory rate from 25 to 35 breaths per minute. Although several ventilatory changes were attempted to compensate for the dead space added by the ACD, effective ventilation could not be achieved. The ACD was removed and placed into the inspiratory limb of the ventilator circuit. This allowed effective ventilation and delivery of isoflurane; however, the authors commented that by using the ACD in this position, there would be a loss of the rebreathing function of the device and no conservation or scavenging of isoflurane. There was also an increase in the dead space of the system. The authors subsequently cautioned against the use of this device distal to the Y-piece in patients weighing less than 30 kg until a smaller model becomes available. They suggested that using the device in the inspiratory limb was a simpler technique than bringing a vaporizer into the PICU setting.
Benzodiazepines
The benzodiazepines remain the most commonly used agent for sedation during mechanical ventilation in the PICU patient. These agents produce amnesia, anxiolysis, and sedation through their effects on the inhibitory neurotransmitter, γ(gamma)-aminobutyric acid (GABA). Benzodiazepines bind to the α(alpha)-subunit of the GABA receptor thereby facilitating binding of the GABA molecule to the β(beta)-subunit resulting in increased chloride conduction across the neuronal membrane and hyperpolarization. Benzodiazepines in common clinical use for sedation in the PICU include midazolam and lorazepam.
Another benzodiazepine, although formerly a commonly used agent, is no longer used in the majority of pediatric ICUs across the country. Although its lipid solubility is greater than that of midazolam, thereby resulting in a more rapid penetration into the CNS and a more rapid onset, its low water solubility requires administration in a solution of propylene glycol. This diluent frequently results in pain and thrombophlebitis when administered through a peripheral vein. Alternatively, diazepam is available in a lipid-based formulation, which has been shown to alleviate the discomfort associated with the intravenous administration of the propylene glycol preparation [73, 74]. More importantly, diazepam is no longer used in the PICU because of its prolonged duration of action with repeated or continuous administration as well as its metabolism to active compounds including oxazepam and N-desmethyldiazepam. These active metabolites have elimination half-lives that far exceed the parent compound.
Midazolam is an imidazobenzodiazepine with a rapid onset of action and a short elimination half-life [75]. Given its rapid onset and water solubility with limited pain on injection, midazolam has found a role for both procedural sedation when administered by intermittent bolus dosing and sedation during mechanical ventilation when used as a continuous infusion except for brief procedures. Clinical experience and years of its use have demonstrated its efficacy for sedation in the PICU patient in doses ranging from 0.05 to 0.2 mg/kg/h [76, 77]. Although previously cost issues were significant, its availability in generic form makes it a cost-effective form of sedation. In a retrospective cohort of 55 pediatric patients requiring mechanical ventilation, sedation was provided by midazolam starting with a bolus dose of 0.25 mg/kg followed by a continuous infusion of 0.4–4 μ(mu)g/kg/min (0.02–0.2 mg/kg/h) [78]. Midazolam was effective in all patients without significant hemodynamic effects. Sedation was ineffective in one patient following the institution of extracorporeal membrane oxygenation (ECMO), which was postulated to be the result of midazolam binding to the membrane oxygenator. Similar efficacy has been reported by other investigators [79]. In the latter study, the investigators noted a significant variation of the plasma concentrations between the patients despite using the same infusion rate. Two infants with gestational ages less than 32 weeks had plasma concentrations greater than 1,000 ng/mL, thereby suggesting decreased clearance in preterm infants. This demonstrates the significant variability in the pharmacokinetics of medications that undergo hepatic metabolism given the immaturity of the hepatic microsomal P450 system.
Although intravenous administration is generally the route chosen in the PICU patient, midazolam remains unique among many of the other agents used for sedation in the ICU setting in that alternative, non-intravenous routes of delivery have been used clinically including oral, rectal, transmucosal (nasal, rectal, sublingual), and subcutaneous administration [80–83]. These non-intravenous routes (oral, rectal, and transmucosal) have generally been used in the arena of procedural sedation or as a premedicant prior to anesthetic induction, while subcutaneous administration has been used with a slow weaning protocol to prevent withdrawal following prolonged administration of benzodiazepines [84]. With all of these non-intravenous routes except for subcutaneous administration, increased doses are required due to decreased bioavailability. Bioavailability is approximately 60–70 % for nasal administration and as low as 30 % for oral administration.
In many centers in the United States, oral midazolam is currently the preferred agent for premedication in the operating room. Doses for oral administration have ranged from 0.25 up to 0.7 mg/kg. If the standard intravenous preparation is used for oral administration, the primary disadvantage is a bitter taste related to the preservative, benzyl alcohol. Although the taste can be masked by mixing the medication in some type of flavored solution, concern has been raised regarding the potential for the alteration of its absorption characteristics. Midazolam normally exists in an equilibrium of two chemical structures: an open and a closed ring form. The ratio of these two compounds in the solution is pH dependent so that with a lower pH, there is more of the open ring or the physiologically inactive configuration. A commercially available preparation of midazolam in a cherry-flavored solution for oral administration is available (Versed syrup, Roche Laboratories Inc, Nutley, NJ). Because of the control of pH during the manufacturing process, clinical data suggest that effective sedation can be achieved with doses as low as 0.25 mg/kg compared to the 0.5–1.0 mg/kg doses reported when using the IV preparation diluted in other solutions for oral administration [85].
Intranasal and sublingual administration has also been well described in the literature. With these routes, as the bioavailability is greater than with oral administration, the dose (0.2–0.4 mg/kg) is lower and the onset more rapid, occurring in as little as 5–10 min. This is avoided with sublingual administration, but issues of taste and patient cooperation may limit the usefulness of this route. With intranasal administration, the patient may object as the preservative, benzyl alcohol, irritates the nasal mucosa. Although the medication is frequently dripped into the nose from a tuberculin syringe, several devices (atomizers) are available that provide a fine mist to contact the mucosa. These may improve contact of the medication with the nasal mucosa, thereby increasing the absorption as well as decreasing the pain related to the diluent.
Midazolam is metabolized by isoforms of the hepatic P4503A enzyme system to the major hydroxylated metabolite, 1-OH midazolam. 1-OH midazolam is approximately equipotent with the parent compound. It undergoes further hepatic metabolism via the glucuronyl transferase system to 1-OH midazolam glucuronide, a water-soluble metabolite, which is renally excreted. In the presence of renal insufficiency, 1-OH midazolam glucuronide accumulates, thereby potentiating the effects of midazolam [86]. Several factors, including age and underlying illness, may also alter midazolam pharmacokinetics. With metabolism dependent on the hepatic P450 system, clearance changes from infancy to adult age and with alterations in hepatic function [87, 88]. Additional changes may occur related to critical illness. In a cohort of 21 PICU patients, midazolam clearance was significantly longer (5.5 ± 3.5 h) than that reported in healthy age-matched children (1.2 ± 0.3 h) [89, 90]. Another issue of clinical significance when continuous infusions are used is the context-sensitive half-time of the medication. Given the pharmacodynamics of many medications, although their half-lives are short and their clinical effects dissipate rapidly following a single bolus dose, the effects can be prolonged following a continuous infusion. This is especially true of drugs that follow a multi-compartmental model [91]. Given these concerns, there is a new benzodiazepine on the horizon, remimazolam, which, like remifentanil, undergoes ester metabolism with a plasma half-life of minutes [92]. Given this unique property, there may be limited context sensitivity in its pharmacokinetic parameters.
Lorazepam is a water-soluble benzodiazepine that is metabolized by glucuronyl transferase to pharmacologically inactive compounds. Medications known to alter the P450 system (anticonvulsants, rifampin, cimetidine) and genetic polymorphisms do not impact lorazepam’s pharmacokinetics. In advanced liver disease, phase II reactions (glucuronyl transferase) are better preserved than phase I reactions (P450 system) so that the pharmacokinetics of lorazepam remain unchanged. Although the Society of Critical Care Medicine’s initial guidelines for sedation of adult patients in the ICU setting recommended lorazepam as the preferred sedative, the most recent guidelines, which were published in 2013, have suggested that sedation with non-benzodiazepines (propofol or dexmedetomidine) may be preferred over lorazepam or midazolam [93, 94]. The guidelines relate this change in practice to the increasing literature suggesting that clinical outcomes are improved with non-benzodiazepine sedation including length of mechanical ventilation and the incidence of delirium.
In comparison to midazolam, there are a limited number of reports regarding the use of lorazepam for sedation in the PICU patient. When comparing lorazepam with midazolam in adult ICU patients, the mean infusion rates to achieve adequate sedation were 0.06 mg/kg/h for lorazepam and 0.15 mg/kg/h for midazolam [95, 96]. There were fewer infusion rate adjustments per day with lorazepam than with midazolam (1.9 for lorazepam versus 3.6 for midazolam). The mean time to return to baseline mental status was also shorter with lorazepam (261 min with lorazepam versus 1,815 min with midazolam). Three of six surviving patients in the midazolam group required more than 24 h to return to their baseline mental status, while all 7 patients in the lorazepam group returned to baseline in less than 12 h.
Prior to its availability as a generic medication, enteral lorazepam was suggested as a means to decrease intravenous midazolam dosing requirements and drug costs during mechanical ventilation in a cohort of 30 infants and children [97]. Midazolam was used for sedation until the requirements were stable for 24 h. Enteral lorazepam equivalence was calculated to be 1/6th of the total daily intravenous midazolam dose. Following the administration of enteral lorazepam, there was a significant reduction in midazolam requirements on day 1, and by day 3, the midazolam infusion was discontinued in 24 of 30 patients. When considering acquisition costs at the time of the study (late 1990s), the projected savings were more than $40,000 for the 30 patients. Enteral lorazepam has also been successfully used for the treatment or prevention of withdrawal following the prolonged administration of intravenous benzodiazepines for sedation during mechanical ventilation in the PICU population [98].
Each mL of the intravenous lorazepam solution (2 mg lorazepam per mL of solution) contains 0.8 mL or 800 mg of propylene glycol. With prolonged or high-dose intravenous administration, issues may arise related to the diluent used in the intravenous formulations, propylene glycol [99–101]. Signs and symptoms of propylene glycol toxicity include metabolic acidosis, renal failure/insufficiency, mental status changes, hemolysis, and an elevated osmolar gap (see later). Propylene glycol is metabolized in the liver to lactic acid and pyruvic acid, which, in part, accounts for lactic acidosis. Propylene glycol is also excreted unchanged in the urine, making toxicity more likely in patients with renal insufficiency. Attention to the propylene glycol infusion rate and periodic calculation of the osmolar gap (measured minus calculated serum osmolarity) may be indicated during high-dose or prolonged lorazepam infusions. An increasing osmolar gap has been shown to be predictive of increasing serum propylene glycol levels [101]. The osmolar gap is used to predict propylene glycol levels as actual plasma propylene glycol concentrations—although they can be measured by reference laboratories (concentrations greater than 18 mg/dL can be associated with toxicity)—are not routinely available in most hospitals. As neonates and especially preterm infants are unable to handle propylene glycol related to hepatic and renal immaturity, continuous infusions of lorazepam are not recommended in this population. In a cohort of 11 PICU patients, who received lorazepam infusions ranging from 0.1 to 0.33 mg/kg/h for 3–14 days, the propylene glycol concentration increased from 86 ± 93 μ(mu)g/mL at baseline to 763 ± 660 μ(mu)g/mL at the completion of the infusion [102]. The plasma propylene glycol concentration correlated with the cumulative dose of lorazepam. No end-organ effects, related to the increased propylene glycol concentrations such as acidosis or hyperosmolarity, were noted in these patients. The authors recommended periodic monitoring for lactic acidosis and hyperosmolarity during prolonged lorazepam infusions.
Etomidate
Etomidate (Amidate™, Abbott Pharmaceuticals) is an intravenous anesthetic agent, introduced into clinical practice in 1972. Its primary effects of sedation and amnesia are mediated through the GABA inhibitory neurotransmitter system. Unlike other sedative and hypnotic agents, only the R(+) enantiomer has clinical effects. Following intravenous administration, loss of consciousness is rapid (15–20 s), and as with propofol and the barbiturates, its duration of action following a single bolus dose is related to redistribution rather than metabolism and clearance. Etomidate undergoes hepatic metabolism with an elimination half-life that varies from 2.9 to 5.3 h [103]. Beneficial CNS effects include a decrease of the cerebral metabolic rate for oxygen (CMRO2), cerebral blood flow (CBF), and ICP. Given its limited impact on myocardial function and blood pressure, cerebral perfusion pressure (CPP) is maintained even in patients with comorbid hemodynamic conditions. In an animal model, no hemodynamic changes were noted with etomidate (0.3 mg/kg), while propofol (2.5 mg/kg) decreased systolic blood pressure by 19.9 %, diastolic blood pressure by 25.3 %, cardiac output by 17.3 %, and systemic vascular resistance (SVR) by 11.6 % [104].
When compared with other sedative agents, there remains limited data regarding the use of etomidate in pediatric-aged patients with that observation that doses of 0.3 mg/kg are required for anesthetic induction [105, 106]. Other investigators found no clinically significant effects following anesthetic induction with etomidate (0.3 mg/kg) in a cohort of 30 pediatric patients with congenital heart disease (left-to-right and right-to-left shunts) [107]. Anecdotally, the successful use of etomidate has been reported in infants and children with depressed myocardial function [108, 109]. Despite the relatively limited clinical and outcome data, recent reviews continue to suggest its use as a single bolus dose for critically ill pediatric patients requiring endotracheal intubation [110, 111]. However, concerns remain regarding the universal use of etomidate for endotracheal intubation in the critically ill pediatric patient (see later).
Etomidate results in a dose-dependent depressant effect on respiratory function and can result in apnea depending on the dose used, concomitant use of other medications, and the patient’s underlying status [112–115]. The effect on ventilator function has been shown to be similar to that which occurs with benzodiazepines and droperidol and less than that with the barbiturates. Etomidate results a shift of the CO2 response curve to the right without a change in the slope. These effects have led some to suggest that etomidate can be used when maintenance of spontaneous ventilation is desirable. However, caution should be maintained; the effect is magnified by the concomitant administration of other sedative and analgesic agents including opioids of benzodiazepines.
Etomidate’s place as an agent for procedural sedation (especially endotracheal intubation) results from its negligible effects on myocardial function, even in patients with significant alterations in myocardial function. This combined with its beneficial effects on the CNS (reduction of the CMRO2 leading to cerebral vasoconstriction, decreased CBF, and decreased ICP) results in maintenance of CPP [116, 117]. Despite its ability to lower CBF and ICP, induction or sedative doses of etomidate can produce increased EEG activity and epileptic-like EEG potentials in patients with underlying seizure disorders [117–120]. Myoclonic movements are also a frequently observed effect following the rapid intravenous administration of etomidate [121]. Although these movements may simulate tonic–clonic seizure activity, no epileptiform discharges are noted. It has been suggested that the myoclonic movements are of spinal origin resulting from disinhibition of inhibitory neuronal pathways. Pretreatment with fentanyl, benzodiazepines, or a small dose of etomidate has been shown to be effective in decreasing the incidence of myoclonus. A trial of etomidate for sedation during computerized tomography was discontinued due to an unacceptably high incidence of involuntary motor movements preventing completion of the scan [122].
The most significant concern with etomidate and the factor that limits its long-term administration in the ICU and perioperative setting are its effects on the endogenous production of corticosteroids. This effect was initially identified when an increased risk of mortality was noted in adult ICU patients who were sedated with a continuous infusion of etomidate [123]. Etomidate inhibits the enzyme 11-β(beta)-hydroxylase which is necessary for a key step in the production of cortisol, aldosterone, and corticosterone. Although the implications of this effect are evident with prolonged infusions or repeated doses, there remains controversy regarding the clinical significance of adrenal suppression following a single dose. However, there are some authorities and investigators calling for the abandonment, or at least a reevaluation, of the use of etomidate [124–127]. The duration of adrenal suppression produced by a single induction dose of etomidate has varied from study to study. Duthie et al. demonstrated a decrease in plasma cortisol levels 1 h following an induction dose of etomidate; however, at 24 h no difference was noted between those patients receiving etomidate and those receiving other induction agents [128]. Other authors have suggested a more prolonged suppression of adrenocortical function. Following a single induction dose (0.3 mg/kg) in pediatric patients undergoing surgery for congenital heart disease, plasma cortisol levels were depressed during cardiopulmonary bypass (CPB), at the end of the operation, and at 24 h [129]. A similar effect with ongoing suppression of adrenal function at 24 h was reported in a cohort of critically ill adult patients [130]. The incidence of adrenal insufficiency (defined as a failure of the serum cortisol level to increase by 9 μ(mu)g/dL after a 250-μ(mu)g ACTH stimulation test) following a single dose of etomidate was 80 % at 12 h, 9 % at 48 h, and 7 % at 72 h [131]. Despite these findings, no difference in outcome was reported following etomidate administration even when there was accompanying effects on adrenal function, and, in fact, vasopressor therapy was required less frequently and in smaller doses when etomidate was used in a cohort of 159 adult patients with septic shock [132].
However, conflicting and more compelling data against the use of etomidate, at least in patients with possible sepsis, comes from the CORTICUS trial [133]. Although the trail was intended to evaluate the efficacy of corticosteroid therapy on outcome in adults with septic shock and adrenal insufficiency, post hoc analysis revealed that patients who had received etomidate had a significantly higher mortality rate. Additionally, this increased risk of mortality was not prevented by the administration of corticosteroids. These data suggest that etomidate should be avoided in patients with sepsis or septic shock; however, there are no definitive data to suggest its elimination from clinical use in other scenarios. Although the use of etomidate has decreased in many centers or has even been totally eliminated, given its beneficial effects on hemodynamic function and intracranial dynamics, until further data are available, it seems prudent to consider its use in critically ill patients outside of the sepsis arena. Perhaps the greater risk may be the potential for cardiovascular collapse when other agents that have significant cardiovascular effects are used in critically ill patients.
In addition to its effects on adrenal function, reports regarding continuous etomidate infusions with increased mortality suggested an association with infectious complications. Neutrophils incubated in vitro with etomidate demonstrate depressed chemiluminescence, an index of oxygen free radical generation, suggesting that etomidate may interfere with white blood cell bactericidal activity [134]. Even without effects on cortisol production, it has been suggested that the incidence of hospital-acquired pneumonia in adult trauma patients may be higher when etomidate is used. In a subset analysis of the HYPOLYTE study, a multicenter trial evaluating the use of hydrocortisone in trauma patients, 95 of the 149 patients enrolled in the trial received etomidate within 36 h of inclusion [135]. Forty-nine (51.6 %) patients who received etomidate developed hospital-acquired pneumonia compared to 16 patients (29.6 %) who did not receive etomidate (p = 0.009). The hazard ratio for hospital-acquired pneumonia was 2.48 with etomidate, although there was no difference in the duration of mechanical ventilation.
Additional reported adverse effects with etomidate, related to the drug itself or the diluent, include anaphylactoid reactions, pain on injection, and an increased incidence of nausea and vomiting [136]. Issues related to the carrier vehicle (propylene glycol) include pain on injection, thrombophlebitis, and propylene glycol toxicity. The incidence of pain on injection has been reported to be as high as 50 %, is greater with injection into small veins on the dorsum of the head, and can be decreased by the preadministration of lidocaine (1.5 mg/kg) or fentanyl (2–3 μ(mu)g/kg). Olesen et al. evaluated the occurrence of pain on injection and the subsequent development of thrombophlebitis in 61 patients randomized to anesthetic induction with either etomidate or thiopentone [137]. Of the patients who received thiopentone, none complained of pain and 4 % developed thrombophlebitis. With etomidate, pain and the development of thrombophlebitis were noted in 24 %. A newer formulation, which contains etomidate dissolved in a fat emulsion of medium- and long-chain triglycerides, may limit the occurrence of injection pain and thrombophlebitis [138]. As with lorazepam, issues may arise with repeated dosing or continuous infusions of etomidate because of the diluent, propylene glycol (please note that given concerns regarding adrenal suppression, long-term etomidate infusions are no longer used in the ICU setting) [139–141].
Despite these issues, given its beneficial effects on CNS dynamics and myocardial function, etomidate has yet to be abandoned in critically ill patients and may still play a role as an effective agent to provide sedation and amnesia during endotracheal intubation [142]. Its lack of cardiovascular effects makes it particularly valuable in patients who may not tolerate a decrease in SVR or myocardial contractility. Given its effects on cerebral dynamics, it also should be considered for patients with increased ICP with or without associated myocardial dysfunction. Given the ongoing concerns regarding the potential for adrenal suppression, ongoing pharmacologic investigation has focused on the development of etomidate-like medications that lack the depressant effect on adrenal function. Carboetomidate was developed based on the hypothesis that the basic nitrogen in the imidazole ring of etomidate binds with high affinity to the heme site of the 11 β(beta)-hydroxylase enzyme [143]. This binding triggers a conformational transition in which the enzyme closes tightly around the bound ligand and becomes nonfunctional. Future clinical trials are needed to fully demonstrate the clinical applications and physiological properties of carbo-etomidate.
Ketamine
Ketamine was introduced into clinical practice during the 1960s [144]. The term dissociative anesthesia is used to describe the state of amnesia and analgesia that is achieved as patients may keep their eyes open and yet be unresponsive to painful stimuli. Ketamine’s anesthetic (sedative, analgesic, and amnestic) properties are mediated through various postulated mechanisms including agonism at opioid receptors and antagonism of NMDA receptors. A unique attribute of ketamine, which separates it from the majority of other agents, is the provision of both amnesia and analgesia. Ketamine contains a chiral carbon in its structure, and the preparation currently used most commonly in clinical practice is a racemic mixture of the two optical isomers S(+) and R(−). However, in the United Kingdom and Europe, the enantiomer S(+) ketamine is available, with the suggestion from preliminary clinical trials that it may provide effective analgesia and sedation while limiting adverse effects including emergence phenomena (see later). Metabolism of ketamine occurs primarily by hepatic N-methylation to norketamine, which retains approximately one-third of the analgesic and sedative properties of the parent compound. Given its dependence on hepatic metabolism, doses should be adjusted in patients with hepatic dysfunction. Dose adjustments may also be required in patients with renal dysfunction since norketamine is dependent on renal elimination.
Beneficial properties of ketamine include preservation of cardiovascular function, limited effects on respiratory mechanics, and maintenance of central control of respiration. These properties make it an effective and popular agent in the arena of procedural sedation during painful, invasive procedures in the spontaneously breathing patient [145]. Incremental doses (0.5–1 mg/kg) can be administered every 1–2 min and titrated to achieve the desired level of sedation and analgesia while generally maintaining spontaneous ventilation. An anti-sialogogue such as glycopyrrolate to prevent salivation and a benzodiazepine to limit the occurrence of emergence phenomena are frequently used with ketamine.
Given its effects at the opioid and NMDA receptors, there is growing interest in the use of ketamine for the management of acute pain, especially during the postoperative period. When coadministered in low doses during morphine analgesia, ketamine has been shown to reduce postoperative opioid consumption and lower opioid-related adverse effects following major surgical procedures in the adult population [146–149]. As NMDA receptor stimulation may be one factor resulting in the development of tolerance to opioid-induced sedation and analgesia, there is interest in the potential benefits of using a low-dose ketamine infusion to delay tolerance during prolonged ICU infusions of morphine and other opioids. Despite the theoretical rationale behind this practice, to date there are no trials to demonstrate a clinically significant effect of this practice.
Ketamine’s popularity in the arena of procedural sedation, especially painful invasive procedures, relates to its beneficial effects on cardiac and respiratory function. Ketamine generally increases heart rate and blood pressure as well as provides bronchodilation due to the release of endogenous catecholamines [150]. Although the indirect sympathomimetic effects from endogenous catecholamine release generally overshadow ketamine’s direct negative inotropic properties, acting to maintain blood pressure and heart rate, hypotension and even cardiovascular collapse may occur in patients with diminished myocardial contractility as ketamine’s direct negative inotropic properties may predominate when endogenous catecholamine stores have been depleted by stress or chronic illness [151–153].
An issue of potential concern and ongoing controversy regarding ketamine is its effects on PVR [154–157]. As these studies were generally performed without full mechanical ventilation support and control of the PaCO2, it is not possible to determine whether the changes in PVR were directly related to ketamine or a consequence of increases in the PaCO2. Most recently, Williams et al. evaluated the effects of ketamine on PVR during sevoflurane anesthesia (0.5 MAC) and spontaneous ventilation in 15 infants and children with pulmonary hypertension (mean PA pressure ≥25 mmHg, baseline PVR index of 11.3 Woods units) [158]. One-third of the patients had PA pressures that were suprasystemic. Ketamine was administered as a bolus of 2 mg/kg followed by an infusion at 10 μ(mu)g/kg/min. There were no significant changes in mean systemic arterial pressure, SVR index, mean pulmonary artery pressure, PVR index, cardiac index, and PaCO2. The safety of ketamine in patients with congenital heart disease is further evidenced by experience with its use during spontaneous ventilation for sedation during cardiac catheterization [159, 160]. In a prospective evaluation of sedation during cardiac catheterization in infants and children, Lebovic et al. reported less hypotension with ketamine compared with propofol, although the recovery times were significantly longer with ketamine [160].
Ketamine has also been shown to have limited effects on several respiratory parameters including functional residual capacity, minute ventilation, and tidal volume [161, 162]. The release of endogenous catecholamines generally results in improved pulmonary compliance, decreased resistance, and prevention of bronchospasm [163]. These effects generally result in the clinical use of ketamine when endotracheal intubation is required in patients with status asthmaticus. Despite the fact that minute ventilation is maintained, hypercarbia with a rightward shift of the CO2 response curve may occur [164]. However, like any sedative/analgesic/general anesthetic agent, ketamine can result in loss of protective airway reflexes, gastric aspiration, and apnea [165, 166]. These latter issues including the potential for respiratory depression are more likely to be clinically significant when ketamine is administered with opioids. An additional effect that may impact on airway patency and the potential for airway obstruction or laryngospasm is that ketamine may increase oral and airway secretions. These effects have resulted in the common clinical practice of administering an anticholinergic agent such as atropine (0.01 mg/kg) or glycopyrrolate (0.005 mg/kg) with ketamine. However, recent studies demonstrate that the actual risk of hypersalivation is low and that the use of these adjunct medications may be unnecessary, thereby resulting in an ongoing controversy regarding ketamine [167–169]. The incidence of airway problems such as laryngospasm with ketamine is higher when there is a history of an antecedent upper respiratory infection and perhaps in those patients chronically exposed to tobacco smoke. Despite the potential for increased airway secretions, reactivity, and even laryngospasm, ketamine has been used effectively for the sedation of infants during flexible fiberoptic bronchoscopy with the maintenance of spontaneous ventilation [170].
An additional area of controversy surrounding ketamine is its effect on ICP. These effects may be indirect, secondary to changes in PaCO2, or the result of a direct effect on the cerebral vasculature. Clinical work from the 1970s reported that ketamine increased ICP, thereby suggesting that it was contraindicated in patients with altered intracranial compliance [171, 172]. These clinical studies were supported by animal investigations demonstrating that the alterations in ICP resulted from direct cerebral vasodilation, which was mediated through central cholinergic receptors [173, 174]. However, more recent data from both animal and human studies have shown no change or even a decrease in ICP following ketamine [175, 176]. Ketamine in doses of 1.5, 3, or 5 mg/kg decreased ICP when administered to adult head trauma patients who were sedated with propofol and mechanically ventilated to maintain a PaCO2 of 35–38 mmHg [177]. The ICP decreased by 2 ± 0.5, 4 ± 1, and 5 ± 2 mmHg with doses of 1.5, 3, and 5 mg/kg respectively. There was no change in CPP, jugular bulb venous oxygen saturation, or middle cerebral artery blood flow velocity. Similar results were reported by Bourgoin et al. when comparing the effects of sedation with either a combination of ketamine and midazolam or sufentanil and midazolam in 25 adult patients with traumatic brain injury (TBI) [178]. The patients were receiving mechanical ventilation and modest hyperventilation. There were no differences in the number of ICP elevations during the study period. Patients receiving ketamine and midazolam had significantly higher heart rate values on days 3 and 4, decreased fluid requirements on day 1, as well as a trend toward decreased vasopressor use when compared with the sufentanil–midazolam group. Mayberg et al. reported that a 1 mg/kg bolus dose of ketamine (1 mg/kg) decreased ICP from 16 ± 1 to 14 ± 1 mmHg with no change in CPP in adult patients anesthetized with isoflurane and nitrous oxide [179]. An additional potentially beneficial effect of ketamine in patients with CNS trauma is an alteration of transmembrane calcium and magnesium currents through its effects on the NMDA receptor [180].
Another somewhat controversial issue related to the CNS effects of ketamine is its use in patients with an underlying seizure disorder. EEG recordings in children and laboratory animals during ketamine administration demonstrate increased frequency and amplitude with occasional paroxysmal seizure activity [181, 182]. However, no clinical evidence of seizure activity has been reported with ketamine administration. Studies in laboratory animals have demonstrated the anticonvulsant effects of ketamine, and there is at least one clinical report as well as animal data describing its use for the treatment of refractory status epilepticus [183–185].
With everyday clinical use, the adverse effect of ketamine that tends to attract the most attention is its potential to cause emergence phenomena or hallucinations. Because of these concerns, clinical practice generally includes the administration of a benzodiazepine (lorazepam or midazolam) along with or prior to the administration of ketamine. The ketamine solution that is in common clinical use is a racemic mixture of the two optically active enantiomers. The single enantiomer form, S(+) ketamine, has been released outside of the United States for clinical use [186–189]. The initial clinical trials have demonstrated that S(+) ketamine is twice as potent as the racemic formulation and offers the clinical advantages of fewer psychomimetic effects, less salivation, and a shorter recovery time [189].
To date, there are only anecdotal reports involving small case series regarding the use of a ketamine infusion for sedation of the PICU patient during mechanical ventilation [190–192]. The largest series included ten patients, ranging in age from 1 week to 30 months. A ketamine infusion, 1 mg/kg/h in five patients and 2 mg/kg/h in the other five patients, was used to provide sedation and analgesia following cardiac surgery in ten pediatric patients [191]. Supplemental doses of midazolam were administered as needed. The two groups had similar and acceptable levels of sedation. No adverse effects were noted. Ketamine has also been used as a therapeutic agent in pediatric patients with status asthmaticus requiring mechanical ventilation [193]. In a cohort of 17 pediatric patients, ketamine was administered as an intravenous bolus (2 mg/kg) followed by a continuous infusion of 20–60 μ(mu)g/kg/min to 17 pediatric patients without changing their preexisting bronchodilatory regimen. There was a significant increase in the PaO2/FiO2 ratio from 116 ± 55 to 174 ± 82, 269 ± 151, and 248 ± 124 at 1, 8, and 24 h, respectively, after the initiation of the ketamine infusion. There was also an improvement in the dynamic compliance as well as a decrease in the PaCO2 and peak inspiratory pressure.
A final caveat with the use of ketamine either by bolus dosing or continuous infusion is that it is commercially available in three different concentrations (100, 50, and 10 mg/mL). Therefore, inadvertent overdosing or underdosing is possible without careful consideration of its concentration. Although it may never become a first-line agent for sedation in the PICU patient during mechanical ventilation, ketamine may be useful in patients who develop adverse cardiovascular effects with opioids or benzodiazepines, for the provision of sedation with the preservation of spontaneous ventilation when using noninvasive ventilation techniques, in patients with status asthmaticus in whom the release of endogenous catecholamines following ketamine administration may provide some therapeutic impact, in low doses by continuous infusion to delay or prevent the development of tolerance to opioids related to its effects at the NMDA receptor, and during the performance of brief, painful invasive procedures in the spontaneously breathing patient. Alternative, non-intravenous routes of delivery have been reported with ketamine including oral and transmucosal (nasal, rectal) administration [188, 194, 195]. These alternative routes of delivery have been used for one-time dosing of the agent for sedation during a procedure or as a premedicant to anesthetic induction. Additionally, ketamine is occasionally administered via the IM route in uncooperative patients without venous access.
Propofol
Propofol is an alkyl phenol compound (2,6-diisopropylphenol) with general anesthetic properties. Although its chemical structure is distinct from that of other intravenous anesthetic, its mechanism of action is similar as it acts through the GABA system [196]. Propofol facilitates the binding of GABA to membrane-bound receptors thereby increasing chloride conductance. Although propofol was initially introduced into anesthesia practice for the induction and maintenance of anesthesia, its rapid onset and recovery times led to its eventual use for sedation in the ICU setting [197, 198]. When compared with midazolam for sedation in adult patients, propofol has been shown to provide shorter recovery times, improved titration efficiency, reduced posthypnotic obtundation, and faster weaning from mechanical ventilation [199].
Propofol, in a manner similar to the barbiturates and etomidate, decreases cerebral metabolic rate of oxygen (CMRO2) leading to reflex cerebral vasoconstriction and a lowering of ICP [200]. These beneficial effects on cerebral dynamics have been validated in animal studies [201, 202]. However, in some experimental studies, adverse hemodynamic effects of propofol required therapy with the direct-acting vasoconstrictor phenylephrine to maintain the mean arterial blood pressure (MAP). When used in clinical practice, the control of MAP has significant clinical impact on propofol’s effect on the ICP. Although ICP is decreased in the majority of human trials, propofol’s lowering of MAP may result in a decrease of the CPP. In patients with intact autoregulation of CBF, a decrease in CPP leads to reflex cerebral vasodilation to maintain CBF, which can secondarily increase ICP if intracranial compliance is altered. This effect negates the decrease in ICP related to the decrease in CMRO2 induced by propofol. In an evaluation of the effects of propofol (2 mg/kg) on ICP and MAP in 6 adults with an ICP ≥25 mmHg, ICP decreased from a mean of 25 ± 3 mmHg to 11 ± 4 mmHg (p < 0.05); however, the reduction of MAP resulted in a decrease of the CPP from 92 ± 8 mmHg to 50 ± 7 mmHg [203]. Similar results have been reported in adults with TBI or during cerebral aneurysm surgery [204–206]. However, if MAP is maintained at baseline with vasoactive agents, propofol may lower ICP and increase CPP. When propofol (2–4 mg/kg/h) was used for sedation during mechanical ventilation in 10 adult patients with TBI, ICP decreased by a mean of 2.1 mmHg at 2 h and the CPP increased by 9.8 mmHg at 24 h [207]. Additionally, beneficial effects of propofol in brain injury include animal data suggesting a protective effect of propofol in various types of hypoxic–ischemic injury models as well as the preservation of the CBF reactivity to carbon dioxide [208–210].
Although ketamine is generally considered the optimal agent for anesthetic induction in patients with active bronchospasm, both laboratory and clinical data support the beneficial effects of propofol on airway reactivity. When comparing the effects of anesthetic induction with propofol (2.5 mg/kg), etomidate (0.4 mg/kg), or thiopental (5 mg/kg) in 77 adults, respiratory resistance was lower after propofol (8.1 ± 3.4 cm H2O/L/s) compared to thiopental (12.3 ± 7.9 cm H2O/L/s) and etomidate (11.3 ± 5.3 cm H2O/L/s) [211]. Pizov et al. randomized a cohort of asthmatic and non-asthmatic patients to anesthetic induction with thiopental/thiamylal (5 mg/kg), methohexital (1.5 mg/kg), or propofol (2.5 mg) [212]. Following endotracheal intubation, auscultation was performed to evaluate the presence of wheezing. In asthmatic patients, the incidence of wheezing was 45 % with thiopental/thiamylal, 26 % with methohexital, and 0 % with propofol. In non-asthmatic patients, the incidence of wheezing was 16 % with thiopental/thiamylal and 3 % with propofol. Propofol’s beneficial effects on airway reactivity are further supported by animal studies showing attenuation of carbachol-induced airway constriction in canine tracheal smooth muscle and prevention of reflex bronchoconstriction to several provocative agents in isolated guinea pig trachea smooth muscle [213, 214]. However, the specific airway and respiratory effects may not be shared by all preparations of propofol. In both an animal model and a human study, these beneficial were present only with the propofol solution that has EDTA as the preservative and not the newer formulations containing sodium metabisulfite or benzyl alcohol [215, 216].
Propofol’s cardiovascular effects resemble those of the barbiturates with the potential for hypotension from peripheral vasodilation and a negative inotropic effect. These effects are dose dependent and can be accentuated following rapid bolus administration and in patients with compromised cardiovascular function. Peripheral vasodilation may be detrimental in patients with a fixed stroke volume (aortic or mitral stenosis) or in the setting of pulmonary hypertension. The adverse hemodynamic profile of propofol administration can be prevented by the administration of calcium chloride [217]. Additional cardiovascular effects may be caused by augmentation of central vagal tone leading to bradycardia, conduction disturbances, and asystole [218–220]. These effects are more likely with the concomitant administration of other medications known to alter cardiac chronotropic function including fentanyl or succinylcholine.
Various neurological manifestations have been reported with the administration of propofol including opisthotonic posturing, myoclonic movements (especially in children), and movements that may resemble seizure-like activity [221–223]. Myoclonus, opisthotonic posturing, and other movements with propofol have been attributed to propofol’s antagonism at glycine receptors in subcortical structures. To date, there is no formal evidence linking propofol with seizures, and its use in patients with seizures and other neurological disorders is acceptable. In a study evaluating the effects of propofol and thiopental on the surface electroencephalograms of 20 patients undergoing temporal lobe surgery, there was no difference between the two groups in the rate of discharge or extension of the irritative zone [224]. Propofol remains an effective agent for the termination of refractory status epilepticus and remains in various published algorithms regarding recommendations for its treatment [225, 226].
Despite its potential benefits in the ICU setting and its efficacy for providing sedation during mechanical ventilation, the routine use of propofol is not recommended and, in fact, is considered contraindicated by many authorities because of the potential for the development of what has been termed the “propofol infusion syndrome.” First described in 1992, by Parke et al., the disorder includes metabolic acidosis, bradycardia, dysrhythmias, rhabdomyolysis, and fatal cardiac failure [227–229]. Eighteen children in the ICU setting with suspected propofol infusion syndrome were reviewed in a report by Bray [230]. The risk factors in the cohort for the development of the syndrome included propofol administration for ≥48 h or an infusion rate ≥4 mg/kg/h. However, not all patients meeting these risk factor criteria developed problems, suggesting that comorbid diseases or a genetic predisposition may be responsible for the development of the propofol infusion syndrome. Additionally, 13 of the 18 patients were ≤4 years of age while only 1 was ≥10 years of age. Subsequent to the initial reports and the review of Bray et al., the syndrome has been reported in older patients including a 17-year-old adolescent and adults [231–234]. In addition to metabolic acidosis and cardiovascular manifestations, additional clinical findings have included lipemic serum, hepatomegaly, rhabdomyolysis, and hyperkalemia. The suggested treatment includes the immediate discontinuation of the propofol combined with symptomatic treatment of cardiovascular dysfunction and acidosis.
Anecdotal evidence suggests that hemodialysis may be helpful as a therapeutic tool by removing a yet undiagnosed metabolite or toxin. Further study has provided insight into the mechanisms of the propofol infusion syndrome. In a guinea pig cardiomyocyte preparation, propofol has been shown to disrupt mitochondrial function [235]. Biochemical analysis of a 2-year-old boy who developed the propofol infusion syndrome revealed an increase in the concentration of C5-acylcarnitine indicative of inhibition of mitochondrial function at complex II of the respiratory chain and an increased plasma concentration of malonylcarnitine [236]. This latter compound inhibits the transport protein necessary for the movement of long-chain fatty acids into the mitochondria. Hemofiltration was used in the treatment of this patient, which resulted in the reversal of the clinical manifestations and recovery of their patient. Similar findings with an elevated serum concentration of acylcarnitine were reported in a 5-month-old who developed propofol infusion syndrome [237]. Treatment was instituted with charcoal hemoperfusion, which resulted in the resolution of the signs and symptoms. These concerns led to the “Dear Healthcare Provider” letter issued in March 2001 by AstraZeneca (Wilmington, DE), the manufacturers of Diprivan®, one of the commercially available propofol preparations [238]. The letter summarized the results of a prospective clinical trial that compared propofol (a 1 or 2 % solution) to other agents used for PICU sedation. There were 12 (11 %) deaths in the 2 % propofol group, 9 deaths (8 %) in the 1 % propofol group, and 4 deaths (4 %) in the standard sedation group. Although subsequent review did not show a specific pattern to the deaths, there was enough concern that the company issued a letter stating: “propofol is currently not approved for sedation in PICU patients in the United States and should not be used for this purpose.” In the majority of large pediatric centers, these concerns have eliminated the routine use of prolonged propofol infusions for sedation in the PICU.
Although propofol has been used safely and effectively for sedation in small cohorts of PICU patients [239–244], its routine use cannot be recommended. In specific clinical scenarios, propofol is still used as a therapeutic tool in the treatment of refractory status epilepticus or increased ICP. In such cases, intermittent analysis of acid–base status and creatinine phosphokinase is suggested. If a base deficit is noted with an increasing serum lactate, immediate discontinuation of the propofol is recommended. Additionally, the short-term administration of propofol (6–12 h) is still used in many centers to transition from other agents such as fentanyl and midazolam to allow for more rapid awakening for tracheal extubation. Short-term propofol infusions may also have a role in the arena of procedural sedation as a means of providing sedation during non-painful invasive procedures such as radiologic imaging. Given its lack of analgesic effects, additional agents (opioids or ketamine) may be required when invasive procedures are performed. Although rare, when such procedures are long, concern has also been expressed regarding the potential development of the propofol infusion syndrome. In a retrospective review of adult patients who received propofol infusion during radiofrequency ablation for atrial fibrillation or atrial flutter, 13 of 55 patients (24 %) had a base deficit of −2 or more compared to 22 of 267 (8.2 %, p < 0.01) in a comparator group of patients undergoing carotid endarterectomy who did not receive propofol [245].
Additional concerns with propofol regarding its use for procedural sedation in spontaneously ventilating patients include a relatively high incidence of respiratory effects including hypoventilation, upper airway obstruction, and apnea, many of which required bag-mask ventilation or repositioning of the airway [246]. When used for procedural sedation, the depth of sedation, assessed using the bispectral index, may be similar to those achieved during general anesthesia. Reeves et al. demonstrated that the low BIS value achieved in children receiving propofol during the performance of a lumbar puncture or bone marrow aspiration was 29.7 ± 13.7 [247].
As propofol is delivered in a lipid emulsion, there may be allergic reactions, pain on injection, and elevated triglyceride levels or hypercapnia with prolonged infusions [248, 249]. It has been theoretically posted that cross-reactivity may occur in patients with allergies to egg, egg products, soybeans, or soy products. Soy allergy is rarely a systemic disease, more likely one limited to intolerance to the soy protein in the gastrointestinal tract. Additionally, although propofol is a soy-based emulsion, all protein components are removed during the manufacturing process. There is no concern in patients with peanut allergy other than the 5 % cross-reactivity with soy. The bigger question arises regarding the safety of propofol use in a patient with egg allergy. To date, there have been no immunologically validated anaphylactic reactions to propofol in this population. Propofol (Diprivan®) contains egg lecithin, which is derived from the egg yolk. The majority of true egg allergies are related to proteins in the egg white. Egg lecithin is a phospholipid compound, which has not been reported to be the provocative agent in allergic reactions. In fact, the literature confirms the safety of propofol use in patients that have been labeled with “egg allergies” and suggests that it is safe in the majority of egg-allergic patients who do not have a history of egg anaphylaxis [250, 251].
A propofol infusion of 2 mg/kg/h provides approximately 0.5 g/kg/day of fat. To limit the impact of the lipid component, a 2 % solution of propofol (twice the amount of propofol with the same amount of lipid per mL as the 1 % solution) has undergone clinical evaluations [252–256]. Although there were fewer problems with hypertriglyceridemia in patients receiving the 2 % solution, there may be an alteration in propofol’s bioavailability as some of these studies have suggested an increased dose requirement and increased incidence of inadequate sedation when the 2 % solution is used compared with the 1 % solution. Given the concerns regarding the lipid component, its fat content should be considered into daily caloric requirements if prolonged infusions are used.
Pain with the injection of propofol remains a significant complaint especially when small veins on the dorsum of the hands or feet are used. Variable success in decreasing the incidence of pain has been reported with various maneuvers including the preadministration of lidocaine, mixing the lidocaine and propofol in a single solution, mixing the propofol with thiopental, diluting the concentration of the propofol, cooling it prior to bolus administration, or the administration of a small dose of ketamine (0.5 mg/kg) prior to the administration of propofol [257–261]. Since propofol has limited analgesic properties, ketamine and propofol can be administered together to take advantage of the analgesia provided by ketamine and the rapid recovery with propofol. There may also be some advantage to the use of a small dose of ketamine (0.5–1 mg/kg) along with a lower dose of propofol (1 versus 3 mg/kg) to initiate the sedation process as this may limit the respiratory and hemodynamic effects of propofol that frequently occur with the initial bolus dose. One final issue with the lipid component of propofol is its potential to serve as a viable growth media for bacteria with reports of bacteremia and postoperative wound infections linked to extrinsically contaminated propofol [262, 263]. Various preservatives are used in the currently available propofol solutions including disodium EDTA (ethylenediaminetetraacetic acid), sodium metabisulfite, and benzyl alcohol. The addition of these substances has dramatically reduced the incidence of bacterial contamination of the solution; however, strict attention to aseptic technique is mandatory. In clinical practice, there may be subtle yet clinically significant differences in these preparations including differential effects on airway reactivity, which have already been discussed in this chapter [215, 216]. The compatibility of various medications is also different with the various propofol preparations [264]. This is an important issue for pediatric patients in whom intravenous access may be limited when infusions may be administered via connectors attached to the same cannula.
Subtle differences in the anesthetic potency of the preparations have also been reported. Although a retrospective analysis of dose requirements during sedation for MRI demonstrated a decreased potency of the sodium metabisulfite propofol solution when compared to the EDTA solution [265], Fassoulaki et al. demonstrated no difference in the cardiovascular or hypnotic effects of the 2 solutions using BIS monitoring [266]. A theoretical disadvantage of the disodium EDTA preparation when used for prolonged infusions in the ICU setting is the chelation and depletion from the body of essential trace minerals such as zinc. Although there are no formal studies to demonstrate that this is a problem, concerns related to this issue are outlined in the manufacturer’s package insert.
Barbiturates
The barbiturates were first synthesized in 1864 by von Baeyer. Thiopental, a short-acting barbiturate, was first administered for clinical use in 1934. This class of anesthetic agent can be classified according to their chemical structure or their duration of activity. The chemical structures of the barbiturates vary in that their ring structure can contain a sulfur atom (thiobarbiturates such as thiamylal and thiopental) or an oxygen atom (methohexital). The presence of a sulfur instead of an oxygen atom in the ring results in a more rapid onset and a shorter duration of action. Increasing the length of the carbon side chains at position 5 of the ring increases the potency of the compound. Short-acting agents such as methohexital, thiopental, and thiamylal have a clinical duration of action of 5–10 min and are used most commonly as a single bolus dose for the induction of anesthesia. When a more prolonged effect is needed, a continuous infusion may be used to maintain constant plasma levels. Long-acting agents with half-lives of 6–12 h include pentobarbital and phenobarbital. The clinical effects of the short-acting agents dissipate rapidly, related to their redistribution, although their hepatic metabolism may take hours. However, when this is done, the offset time will also be markedly prolonged and dependent on the duration of the infusion. In the PICU setting, the barbiturates are occasionally used by continuous infusion for sedation during mechanical ventilation (see later) although their more common use is based on their beneficial physiologic and therapeutic effects as anticonvulsants or to decrease ICP in patients with TBI [267–272].
Like propofol, the effects of the barbiturates on hemodynamic and respiratory function are dose dependent. In healthy patients, sedative doses have limited effects on cardiovascular function, respiratory drive, and airway protective reflexes, while larger doses, especially in patients with cardiorespiratory compromise, will result in respiratory depression, apnea, or hypotension. Hypotension results from peripheral vasodilation, a direct negative inotropic effect, and blunting of the sympathetic nervous system. On a cellular level, the barbiturates inhibit calcium fluxes across cell membranes and from the sarcoplasmic reticulum thereby depressing myocardial contractility. These effects are magnified in patients with comorbid cardiovascular diseases and in the presence of hypovolemia. These agents should be used cautiously, if at all, in patients with cardiovascular dysfunction. Additionally, the effects on cardiovascular and ventilatory function are additive with other agents such as opioids.
The ultrashort-acting barbiturates (thiopental and thiamylal) are used clinically in a 2.5 % solution with a pH of 10.5. The high pH results in a bacteriostatic solution limiting concerns of bacterial contamination as well as limiting the pain that may occur with intravenous injection. However, a pH of 10.5 leads to incompatibilities with other medications and parenteral alimentation solutions, thereby necessitating a separate infusion site if a continuous infusion is used. Of particular note is the potential for the barbiturates to form precipitates when administered with drugs such as rocuronium, mandating flushing the line during rapid administration of medications during maneuvers such as rapid sequence intubation to avoid loss of intravenous access during critical moments. Local erythema, thrombophlebitis, or skin sloughing may occur with subcutaneous infiltration. The barbiturates possess no analgesic properties and therefore should be used with an opioid in situations requiring analgesia.
The barbiturates’ place in ICU sedation appears to be as an alternative or second-line agent when primary agents, either alone or in combination, fail to provide adequate sedation or result in untoward side effects [273]. There are a limited number of reports regarding the use of pentobarbital infusions for sedation in the PICU setting. A retrospective report described the use of pentobarbital for sedation during mechanical ventilation of 50 infants and children, ranging in age from 1 month to 14 years [274]. A pentobarbital infusion was started when a combination of a benzodiazepine (midazolam in doses of 0.4 mg/kg/h) and an opioid (either fentanyl in doses of 10 μ(mu)g/kg/h or morphine in doses of 100 μ(mu)g/kg/h) did not provide effective sedation. The midazolam infusion was discontinued when the pentobarbital infusion was started. In 12 patients, opioid infusion was continued for more than 48 h after starting the pentobarbital infusion to control pain related to a surgical procedure or an acute medical illness. In the other patients, the opioid infusion was discontinued. Pentobarbital was administered for a median duration of 4 days (range 2–37 days) at a median dose of 2 mg/kg/h (range 1–6 mg/kg/h). Once pentobarbital was started, neuromuscular blocking agents, which had been required in seven patients due to excessive movement and inadequate sedation, were discontinued. Additionally, there was no longer a need for direct-acting vasodilators (sodium nitroprusside or nicardipine) in the five patients who had previously required these agents. The cohort also included seven non-neonatal ECMO patients in whom pentobarbital provided effective sedation. Tolerance was noted with the administration of pentobarbital. In the 14 patients that received pentobarbital for ≥5 days, the dose requirements increased from 1.2 mg/kg/h on day #1 to 3.4 mg/kg/h on day #5. No significant adverse effects related to pentobarbital were noted. Six of the 36 patients who had received pentobarbital for more than 4 days manifested signs and symptoms of withdrawal. An opposing opinion is expressed by Yanay et al. who reported their retrospective experience with pentobarbital sedation for eight PICU patients [275]. Although pentobarbital provided effective sedation and allowed the discontinuation of neuromuscular blocking agents, they noted a relatively high incidence of adverse effects including blood pressure instability (25 %), oversedation (12.5 %), and neurologic sequelae (12.5 %) including withdrawal phenomena. These adverse effects led to the discontinuation of the drug in 25 % of their patients.
In addition to their role for therapeutic agents or perhaps for the provision of sedation during mechanical ventilation, there are several reports outlining the use of various barbiturates for procedural sedation. As they have no intrinsic analgesic properties, the barbiturates are used most commonly for sedation during non-painful procedures. The short-acting oxybarbiturate, methohexital, has been used extensively via both the oral and PR route as a sedative for CT or MR imaging with success rates of up to 80–85 % [276]. The standard dose per rectum is 20–30 mg/kg, which produces a rapid onset of sleep (6–10 min) with recovery to baseline status within 1.5–2 h. Adverse effects are uncommon with mild respiratory depression responsive to repositioning or the administration of supplemental oxygen occurring in up to 4 % of patients. The duration of action with intravenous use (0.75–1.0 mg/kg) is approximately 10 min, making the drug attractive for short procedures such as computerized tomography (CT imaging). However, the incidence of respiratory depression is greater with the intravenous route of administration, which may limit its usefulness. Unlike the other barbiturates, methohexital may activate the electroencephalogram (EEG) and has been reported to precipitate seizures in patients with underlying seizure disorders.
Although used most commonly by the intravenous route for the induction of anesthesia, thiopental has also been used as a rectal agent for sedation for radiologic procedures in doses of 25–50 mg/kg [277, 278]. When compared with methohexital, the depth of sedation achieved and reported success rates were somewhat higher (>90 %). The onset of action is slightly longer (15–30 min) with a similar duration of action (60–90 min) compared to methohexital. Pentobarbital has an intermediate duration of action and remains a popular choice for intravenous sedation during radiologic procedures such as MR imaging where sedation times may approach 60–90 min. Multiple delivery options are available including the IV, IM, and enteral routes, although IV administration remains the most commonly used route. Pentobarbital is administered in increments of 1–2 mg/kg every 3–5 min until sleep is induced (average total dose 4–5 mg/kg) [279, 280]. The average duration of sleep after a single intravenous dose is 60–90 min, which is adequate to perform most routine MRI evaluations. Respiratory depression and hypotension may occur, especially with rapid intravenous administration. Disadvantages with pentobarbital include prolonged recovery times (2–4 h) and emergence issues including agitation.
Opioids
Although generally used for analgesia, opioids also possess sedative properties, especially those with agonistic effects at the κ(kappa) opioid receptor [281]. Therefore, these agents may be effective for providing sedation during mechanical ventilation and remain second to the benzodiazepines as the most commonly used agents in the PICU setting. Although the opioids provide analgesia, even in settings such as anesthesia for cardiovascular surgery when high doses of specific agents (fentanyl 50–75 μ(mu)g/kg) are administered, amnesia is not provided. Therefore, additional agents are required in situations that demand amnesia such as a patient who is receiving a neuromuscular blocking agent. In patients with altered myocardial function or at risk for pulmonary hypertension, such as an infant with a large preoperative left-to-right shunt, particular benefit has been reported with the synthetic opioids as they provide cardiovascular stability, beneficial effects on PVR, and blunting of sympathetic stress response. Due to their prompt redistribution and resultant short plasma half-lives following bolus administration, the synthetic opioids are generally administered by a continuous infusion to maintain plasma concentrations adequate to provide sedation and analgesia.
The synthetic opioids that are currently in common clinical use include fentanyl, sufentanil, and remifentanil. Both fentanyl and sufentanil are dependent on hepatic metabolism, while remifentanil is depending on metabolism by nonspecific esterases (see later). Several clinical scenarios may alter hepatic metabolic function and thereby prolong the half-lives of these agents, including immaturity of the hepatic microsomal enzymes as is seen in term and especially preterm infants, decreased hepatic blood flow that occurs following intra-abdominal procedures, and primary hepatic diseases with hepatocellular dysfunction. Although these agents are short acting when administered as a single bolus dose, they also have a context-sensitive half-life so that the duration of their effect is prolonged when they are administered over an extended period of time. This effect is greater with fentanyl than sufentanil.
Remifentanil is metabolized by nonspecific esterases in the plasma. It has a clinical half-life of 5–10 min and a brief duration of effect even following 12–24 h of continuous infusion thereby not demonstrating changes related to a context-sensitive half-life [282]. These pharmacokinetic parameters hold true even in the neonatal population, making remifentanil the only opioid whose pharmacokinetics are not altered by gestational or chronologic age [283]. Given these properties, it is a potentially useful agent for providing a deep level of sedation and yet allowing for rapid awakening with discontinuation of the infusion even in the neonatal population. Although there is significant clinical experience with the use of remifentanil during surgical procedures in patients of all ages, to date there remains limited experience with its use in the ICU population. Cavaliere et al. evaluated the efficacy of a remifentanil infusion in doses starting at 0.02 μ(mu)g/kg/min and increasing up to 0.25 μ(mu)g/kg/min, in providing sedation during mechanical ventilation in a cohort of ten adult ICU patients [284]. Although sedation, assessed by clinical sedation scales, was adequate in the ten patients, the maximum infusion rate was achieved in only four of the ten patients due to the occurrence of adverse effects including hypotension and bradycardia at infusion rates ≥0.15 μ(mu)g/kg/min. Hypoventilation was noted at infusion rates as low as 0.1 μ(mu)g/kg/min. The authors concluded that low doses of remifentanil (0.05 μ(mu)g/kg/min) provided effective sedation in critically ill patients while the adverse effect profile limited the use of higher doses. In a prospective randomized trial, adults requiring mechanical ventilation received either a morphine infusion at 0.75 μ(mu)g/kg/min or a remifentanil infusion at 0.15 μ(mu)g/kg/min [285]. Although the percentage of optimal sedation hours was significantly greater with remifentanil with fewer dose adjustments, the duration of mechanical ventilation and extubation time was significantly longer with morphine. There was no difference in the incidence of adverse effects.
To date, there are limited data regarding the use of remifentanil for sedation during mechanical ventilation in the PICU population [286, 287]. Our preliminary clinical experience demonstrates that remifentanil may be useful in providing sedation during mechanical ventilation in patients with TBI who require frequent and intermittent neurological examination. In this population, the short half-life of remifentanil allows the successful neurological examination while providing a sedation plan of analgesia and sedation in the interim.
Although these anecdotal reports demonstrate that remifentanil may be an effective agent, providing a deep level of sedation with rapid awakening when the infusion is discontinued, an issue that needs further investigation prior to its widespread application in the ICU setting is the rapid development of tolerance. The development of tolerance has been demonstrated after even brief infusions of less than 60–90 min [288, 289]. This has translated into greater postoperative opioid requirements when remifentanil is used intraoperatively and the need to escalate doses rapidly when remifentanil is used for ICU sedation. Although tolerance may limit prolonged remifentanil infusions, there remains interest in the use of remifentanil in the arena of procedural sedation given that its effects dissipate rapidly when the infusion is discontinued [290–292]. Given its analgesic properties, remifentanil has been combined with midazolam or propofol as a means of providing analgesia during painful, invasive procedures. Despite its efficacy in this arena, reports demonstrate a significant incidence of respiratory depression and apnea, which may limit its applicability in this setting. However, given the ability of the opioids to blunt the cough reflex, remifentanil may have a role during bronchoscopy or when fiberoptic intubation of the trachea is necessary [291].
Two additional issues relevant to the synthetic opioids are potential effects on ICP and the risks of chest wall rigidity. Anecdotal reports suggested the potential for the synthetic opioids to increase ICP and decrease CPP in adults with altered intracranial compliance [293]. Rather than a direct effect, the mechanism responsible for the ICP increase has been shown to be a reflex cerebral vasodilation in response to the decrease in MAP or CPP [294]. A similar effect has been described with propofol (see earlier section). If the CPP or MAP is maintained with a direct-acting vasoconstrictor, no change in ICP is noted thereby making these agents safe and effective in patients with altered intracranial compliance. A second adverse effect specific to the synthetic opioids is chest wall and laryngeal rigidity [295, 296]. These effects are related to the dose and the rate of administration, are centrally mediated responses that can interfere with respiratory function, and their incidence can be decreased by premedication with the α(alpha)2-adrenergic agonists, reversed with naloxone, and interrupted with neuromuscular blocking agents. Although rare, its occurrence should be considered if respiratory dysfunction is noted following the use of synthetic opioids. In severe cases, this effect may interfere with respiratory function resulting in rapid oxygen desaturation and hypoxemia [297].
Given the issues with the rapid development of tolerance following the use of the synthetic opioids, morphine has regained popularity for sedation and analgesia during mechanical ventilation in the PICU setting. As morphine has agonistic effects at both the mu and the kappa opioid receptor, it provides not only analgesia via the mu receptor but also sedation via the kappa receptor. Cardiovascular effects include dilation of the venous capacitance system with a decrease in preload, which may result in a modest decrease in blood pressure, especially in patients with decreased intravascular volume or comorbid cardiac diseases. When used by continuous infusion for sedation during mechanical ventilation in neonates, morphine has been shown to have no effect on intelligence, motor function, or behavior [298].
In infants, morphine infusions of 10–30 μ(mu)g/kg/h provided effective analgesia and sedation during mechanical ventilation after surgery for congenital heart disease without impairing the ability to wean mechanical ventilatory support [299]. Morphine infusions blunt the sympathetic response and reduce epinephrine levels in neonates requiring endotracheal intubation and mechanical ventilation for hyaline membrane disease [300]. In a cohort of infants requiring sedation and analgesia during ECMO (mean duration of ECMO 4–5 days), morphine and fentanyl provided equivalent levels of sedation while decreasing the need for supplemental bolus doses of opioid [301]. Infants receiving morphine had a lower incidence of withdrawal (13 of 27 with fentanyl versus 1 of 11 with morphine, p < 0.01) and were hospitalized for fewer days after ECMO (31.1 ± 14 versus 21.5 ± 7.0 days, p = 0.01).
Although administered most commonly via the intravenous route, rare circumstances, such as limited intravenous access or drug incompatibilities, may occur that preclude intravenous administration in the PICU setting. In such situations, the subcutaneous administration of opioids is feasible. Although used most commonly in the control of chronic cancer pain [302], there is experience with subcutaneous opioid infusions in the ICU setting. Bruera et al. successfully used subcutaneous opioids, administered by intermittent dosing or continuous infusions, in adult ICU patients [303]. The infusions were delivered through a 25 gauge butterfly needle inserted subcutaneously in the subclavicular area or the anterior abdominal wall. No infectious complications were noted and the insertion site was changed only three times due to local problems such as erythema. Although the authors expressed a theoretical concern over possible delays in the onset of activity or decreased absorption in patients with decreased peripheral perfusion, they noted no problems in their cohort.
There is also anecdotal experience with the use of subcutaneous opioids in the PICU population [304–306]. A retrospective evaluation regarding the subcutaneous administration of fentanyl in 24 PICU patients ranging in age from 2 weeks to 18 years demonstrated the efficacy of the technique in the control of postoperative pain, as a gradual weaning regimen following prolonged opioid use, or for the provision of comfort during the terminal stages of a disease [306]. The subcutaneous fentanyl infusions were administered when intravenous administration was not feasible due to lack of intravenous access or drug incompatibilities.
As with all of the previously described agents, opioids may have adverse effects on respiratory function with the potential for hypoventilation or apnea. However, an effect that appears to be relatively specific to the opioids is their potential impact on immune function. Opioid receptors have been found on immune cells that participate in the inflammatory response and various host defenses. Binding of opioids to these receptors decreases inflammation and may play some role in the control of acute pain by opioids. However, in specific circumstances, this effect may be deleterious. Increased viral loads have been noted in patients with HIV infections who are receiving methadone [307]. Opioids modulate cytokine production and in an animal model morphine has been shown to reduce reticuloendothelial cell function, phagocytic count, phagocytic index, killing properties, and superoxide anion production [308, 309]. Although there are no studies directly linking these effects to adverse clinical outcomes, additional studies are needed to define these effects, their mechanisms, and most importantly their impact on the PICU patient.
Phenothiazines and Butyrophenones
The phenothiazines and butyrophenones are classified as the “major tranquilizers.” The majority of their clinical use is in the treatment of psychiatric disturbances or as antiemetics in various clinical scenarios. Of the several agents available, haloperidol is the agent that has been used most frequently for the sedation of adults in the ICU setting. Haloperidol acts through central dopamine receptors. With intravenous administration, its onset of action is within 10–20 min with a duration of action of 12–24 h given its long elimination half-life of 18–26 h [310]. Although not formally approved by the FDA for intravenous administration, there is significant and adequate clinical experience with its use by this route in the adult population [311]. Riker et al. reported their experience with the continuous infusion of haloperidol in doses ranging from 3 to 25 mg/h for sedation in eight adult ICU patients [312]. They proposed various benefits of haloperidol including a rapid onset, minimal respiratory depression, and lack of active metabolites. A retrospective report regarding haloperidol use in a cohort of 989 adult patients, who required mechanical ventilation for more than 48 h, reported not only efficacy in controlling agitation and delirium, but also a lower overall mortality in patients who received haloperidol [313]. Patients who received haloperidol within 48 h of the initiation of mechanical ventilation had a lower inhospital mortality (20.5 % versus 36.1 %, p = 0.004) when compared with those who did not receive haloperidol. These differences persisted even when adjusted for age, comorbid features, severity of illness, degree of organ dysfunction, and admitting diagnosis. Because of the retrospective nature of the study and the potential risks associated with haloperidol (see below), the authors suggested that prospective randomized trials were needed before applying this therapy routinely to the ICU population.
Experience with haloperidol in the PICU population remains anecdotal. Harrison et al. reported their experience with haloperidol, administered by intermittent bolus dosing, in five critically ill children [314]. The patients ranged in age from 9 months to 16 years and had become difficult to sedate despite escalating doses of benzodiazepines and opioids. Haloperidol dosing included a loading dose of 0.025–0.1 mg/kg, repeated every 10 min until the patient was sedated. The total loading dose required ranged from 0.09 to 0.25 mg/kg. This was followed by intermittent doses of 0.015–0.15 mg/kg (daily maintenance dose of 0.06–0.45 mg/kg/day) administered every 8 h. Haloperidol’s efficacy was demonstrated by a reduction of opioid and benzodiazepine requirements, decreased need for supplemental doses of sedative agents, decreased use of neuromuscular blocking agents, and improved clinical sedation. One patient developed a dystonic reaction, which resolved in 36 h without therapy as the haloperidol had already been discontinued.
Potential adverse effects associated with the butyrophenones and phenothiazines include hypotension related to peripheral α(alpha)-adrenergic blockade, dystonic and extrapyramidal effects, lowering of the seizure threshold, the neuroleptic malignant syndrome, and cardiac arrhythmias including torsades de pointes due to effects on cardiac repolarization. In the study of Riker et al., one of the eight adult patients developed atrial dysrhythmias, prolongation of the QT interval, and ventricular tachycardia [312]. The potential for cardiac dysrhythmias due to alterations in repolarization may be exacerbated in critically ill patients with altered sympathetic function related to fever, pain, or the stresses of an acute illness. Similar issues may occur with other drugs of this class including droperidol [315]. Through a black box warning issued by the US Food and Drug Administration, concern has been expressed regarding the potential association of droperidol and postoperative cardiac events including torsades de pointes in adult patients [316]. Prolonged postoperative electrocardiogram (ECG) monitoring is suggested in patients treated with droperidol during the perioperative period.
Alpha2-Adrenergic Agonists
Although used initially for clinical effects such as the control of blood pressure, the α(alpha)2-adrenergic agonists including clonidine and dexmedetomidine may also have a role in the PICU patient for the provision of sedation during mechanical ventilation, reduction of opioid requirements, control of pain of various etiologies, and provision of sedation during noninvasive procedures. The physiologic effects of these agents are mediated via stimulation of postsynaptic α(alpha)2-adrenergic receptors that activate a pertussis toxin-sensitive guanine nucleotide regulatory protein (G protein), resulting in decreased activity of adenylyl cyclase [317, 318]. The subsequent reduction in intracellular cyclic adenosine monophosphate (cAMP) and cAMP-dependent protein kinase activity modifies membrane ion conductance resulting in decreased neuronal activation providing sedation and anxiolysis [319, 320].
Activation of receptors in the medullary vasomotor center reduces norepinephrine turnover and decreases central sympathetic outflow, resulting in alterations in sympathetic function with decreased heart rate and blood pressure. Additional effects result from the central stimulation of parasympathetic outflow and inhibition of sympathetic outflow from the locus ceruleus in the brainstem. The latter effect plays a prominent role in sedation and anxiolysis produced by these agents as decreased noradrenergic output from the locus ceruleus allows for increased firing of inhibitory neurons including the GABA system resulting in sedation and anxiolysis [321]. This effect has been shown to be similar to that which occurs during non-REM sleep [322, 323]. The lack of non-REM sleep with the use of other sedative agents including propofol, benzodiazepines, and barbiturates is one of the factors that may result in delirium in adult ICU patients. This mechanism of action has led to the increased use of this agent in adult ICUs where concerns regarding the impact of delirium on outcome have been emphasized in the recent literature. The α(alpha)2-adrenergic agonists also potentiate the analgesic effects of opioids by regulating substance P release within the central nervous system [324].
Clonidine has been used as a premedicant in the operating room, for caudal and epidural analgesia, as an adjunct to opioid-induced analgesia during the postoperative period, and even for ICU sedation [325–328]. Although initially available only as a tablet, clonidine is now available as a transdermal patch and as a preparation for neuraxial administration. The latter has been administered intravenously in various clinical scenarios. In an open label evaluation in children in the PICU setting, a continuous clonidine infusion starting at 1 μ(mu)g/kg/min was added to a continuous midazolam infusion of 1 μ(mu)g/kg/min [328]. No significant change in heart rate, blood pressure, or cardiac index was noted. In 2 of the 20 patients, the clonidine infusion was increased to 2 μ(mu)g/kg/h. The clonidine infusion provided adequate sedation for 602 of the 672 study hours with no sedation failures. Arenas-Lopez et al. reported their experience with the addition of enteral clonidine (3–5 μ(mu)/kg every 8 h) as an adjunct to intermittent doses of morphine and lorazepam for sedation during mechanical ventilation in 14 children [329]. Adequate sedation was achieved during 82 % of the study period with an overall decrease in the requirements for both lorazepam and morphine. No adverse effects were noted. With the increased use of and accumulating clinical experience with dexmedetomidine, the use of clonidine has diminished in most pediatric ICUs [330].
Like clonidine, dexmedetomidine is a centrally acting, α(alpha)2-adrenergic agonist and exhibits the same physiologic effects. However, it possesses an affinity eight times that of clonidine for the α(alpha)2-adrenergic receptor, a differential α(alpha)1 to α(alpha)2 agonism of 1:1,600, and a half-life of 2–3 h thereby allowing its titration by intravenous administration. In healthy adult volunteers, the pharmacokinetic profile of dexmedetomidine includes a rapid distribution phase with a distribution half-life of approximately 6 min, an elimination half-life of 2 h, and a steady-state volume of distribution of approximately 118 L. Dexmedetomidine exhibits linear kinetics, is 94 % protein bound, and undergoes hepatic metabolism with minimal unchanged drug excreted in the urine and feces. Given its dependence on hepatic metabolism, dose adjustments are necessary in patients with altered hepatic function.
The pharmacokinetic profile of dexmedetomidine has been well studied in the pediatric population and in various clinical scenarios [331–336]. Diaz et al. studied the pharmacokinetics of dexmedetomidine in a cohort of 10 children ranging in age from 0.3 to 7.9 years following cardiac (n = 9) or craniofacial procedures (n = 1) [332]. Dexmedetomidine was administered as a bolus dose of 1 μ(mu)g/kg followed by an infusion of 0.2–0.7 μ(mu)g/kg/h for 8–24 h (median duration of administration of 19.6 h). In this cohort, the authors noted pharmacokinetics similar to those reported in the adult population and pediatric patients without CHD. Using a two-compartment model, the volume of distribution was 1.53 ± 0.37 L/kg, the clearance was 0.57 ± 0.14 L/kg/h (approximately 9.5 mL/kg/min), and the terminal elimination half-life was 2.65 ± 0.88 h. They also noted a low interpatient variability (coefficient of variation of 25 %) in their cohort of ten pediatric patients. Based on the clearance data from their study, it was determined that an infusion rate of 0.35 μ(mu)g/kg/h (range: 0.25–0.47 μ(mu)g/kg/h) should achieve a therapeutic plasma concentration of 600 pg/mL. Based on experience with several other pharmacologic agents, they cautioned that the pharmacokinetics of dexmedetomidine is likely to be significantly different in patients less than 4 months of age.
Potts et al. and Su et al. provide additional data regarding dexmedetomidine pharmacokinetics in the pediatric cardiac surgical population [334, 335]. The first of these studies administered a single bolus of dexmedetomidine over 10 min in a dose varying from 1 to 4 μ(mu)g/kg to a cohort of 45 pediatric patients, ranging in age from 4 days to 14 years [334]. As with previous investigations, the authors found that the pharmacokinetics best fits a two-compartment model. Clearance at birth was 15.55 L/h per 70 kg and matured with a half-time of 46.5 weeks to reach 87 % of adult values by 1 year of age. In distinction to other studies, no age-related changes in the volume of distribution were noted. Simulation of a bolus of 1 μ(mu)g/kg followed by an infusion of 0.7 μ(mu)g/kg/h suggested that children arouse from sedation at a plasma concentration of 0.304 μ(mu)g/mL (the suggested therapeutic level for sedation in adults is 0.600 μ(mu)g/mL). No comment was made regarding differences based on the type of congenital heart disease (single versus two ventricle, cyanotic versus non-cyanotic). Su et al. provide more specific information regarding the impact of CPB and the type of CHD on the pharmacokinetics of dexmedetomidine [335]. Sequential cohorts of 12 infants each who required mechanical ventilation following surgery for CHD were enrolled to receive one of three dosing regimens. The three regimens (loading dose and continuous infusion) included 0.35–0.25, 0.7–0.5, and 1–0.75 μ(mu)g/kg and μ(mu)g/kg/h respectively. The authors noted that dexmedetomidine clearance increased with weight, age, and single-ventricle physiology, while total CPB time was associated with a trend toward decreased clearance. Potts et al. performed a pooled analysis of four of the studies from the pediatric population in an attempt to provide a summary of the pharmacokinetics of dexmedetomidine in the pediatric-aged patient [336].
The effects of dexmedetomidine on sedation, hemodynamic and respiratory function were evaluated in a cohort of healthy adults [337]. The volunteers received either a bolus of saline followed by a saline infusion or 0.6 μ(mu)g/kg of dexmedetomidine infused over 10 min followed by a dexmedetomidine infusion at either 0.2 or 0.6 μ(mu)g/kg for 1 h. The level of sedation was graded by the patient (VAS level of sedation from 0 = asleep to 100 = wide awake), an observer, and by using the BIS monitor. The two dexmedetomidine infusions resulted in similar and significant degrees of sedation with limited changes in hemodynamic and respiratory function. In addition to its sedative properties, dexmedetomidine has been shown to decrease opioid requirements following surgical procedures. When compared with placebo in 119 adult patients who required mechanical ventilation following cardiac and general surgical procedures, patients receiving dexmedetomidine required 80 % less midazolam and 50 % less morphine [338]. Dexmedetomidine dosing included an initial bolus dose of 1 μ(mu)g/kg followed by an infusion of 0.2–0.7 μ(mu)g/kg/h. Eighteen of the 66 patients who received dexmedetomidine experienced hypotension (MAP less than 60 mmHg or a greater than 30 % decrease from baseline) or bradycardia (heart rate less than 50 beats per minute). The cardiovascular changes were noted during the bolus dose in 11 of the 18 patients.
To date, there remains only one prospective trial evaluating dexmedetomidine for sedation during mechanical ventilation in pediatric-aged patients [339]. Efficacy was evaluated using the Ramsay scale and by comparing the requirements for supplemental morphine. Dexmedetomidine at 0.25 μ(mu)g/kg/h provided sedation that was equivalent to midazolam at 0.22 mg/kg/h. Dexmedetomidine at 0.5 μ(mu)g/kg/h was more effective than midazolam, as demonstrated by a decreased need for supplemental morphine and a decrease in the number of Ramsay scores of 1 exhibited by the patients. Dexmedetomidine was somewhat less effective for patients ≤12 months of age as five of the six patients that exhibited a Ramsay score of 1 during dexmedetomidine were less than 12 months of age. The only adverse effect was bradycardia in one patient receiving dexmedetomidine who was also receiving digoxin [340].
In addition to its use for sedation during mechanical ventilation, other applications of dexmedetomidine have included procedural sedation, prevention of shivering, and treatment of iatrogenic opioid and benzodiazepine withdrawal following prolonged use in the ICU setting [336]. Koroglu et al. randomized 80 children (1–7 years of age) to dexmedetomidine or midazolam during MR imaging [341]. Dexmedetomidine was administered as a loading dose of 1 μ(mu)g/kg over 10 min followed by an infusion of 0.5 μ(mu)g/kg/h, while midazolam was administered as a loading dose of 0.2 mg/kg followed by an infusion of 6 μ(mu)g/kg/h. The quality of sedation was better and the need for rescue sedation was less (8 of 40 versus 32 of 40) with dexmedetomidine compared to midazolam. Similar efficacy was reported in an open label trial of dexmedetomidine for sedation during magnetic resonance (MR) imaging in 48 pediatric patients ranging in age from 5 months to 16 years, 15 of whom had failed sedation with another agent [342]. A second study by Koroglu et al. randomized 60 children to dexmedetomidine or propofol during MR imaging [343]. Although both of the agents were equally effective in providing sedation, propofol provided shorter induction times, recovery times, and discharge times. However, adverse effects including hypotension and oxygen desaturation were more common with propofol. Oxygen desaturation requiring intervention including a chin lift, discontinuation of the infusion, or supplemental oxygen occurred in 4 of 30 children receiving propofol versus 0 of 30 receiving dexmedetomidine.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


