Fifth World Symposium on Pulmonary Hypertension: Nice, France, 2013
1. Pulmonary arterial hypertension
Idiopathic PAH
Heritable PAH
Drug and toxin induced
Associated with
Connective tissue diseases
HIV
Portal hypertension
Congenital heart disease
Schistosomiasis
Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis
Persistent pulmonary hypertension of the newborn
2. Pulmonary hypertension because of left heart disease
Left ventricular systolic dysfunction
Left ventricular diastolic dysfunction
Valvular disease
Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies
3. Pulmonary hypertension because of lung disease and/or hypoxia
Chronic obstructive pulmonary disease
Interstitial lung disease
Other pulmonary diseases with mixed restrictive and obstructive pattern
Sleep-disordered breathing
Alveolar hypoventilation syndrome
Chronic exposure to high altitude
Developmental abnormalities
4. Chronic thromboembolic pulmonary hypertension
5. Pulmonary hypertension with unclear multifactorial mechanisms
Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy
Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
Metabolic disorders: glycogen storage disorders, Gaucher disease, thyroid disorders
Others: tumor obstruction, fibrosing mediastinitis, chronic renal failure
Abbreviations: HIV, human immunodeficiency virus; PAH, pulmonary arterial hypertension.
CLINICAL PRESENTATION
Two clinical scenarios have been observed regarding PAH and pregnancy that involve patients diagnosed before pregnancy and those diagnosed during pregnancy. In the former group, both patient and clinician are aware of the precarious impact of pregnancy on the mother as many have been warned to avoid pregnancy and informed of the hazardous effect of PAH medications on the fetus, in particular, the endothelin receptor antagonists (Table 11-2). Pregnancy may be unexpected because of lack of birth control or contraceptive failure, while other patients become pregnant because of the misperception that medical therapies will allow them to have an uncomplicated and safe delivery. In contrast, patients without a prior diagnosis tend to be diagnosed later in pregnancy as their symptoms are misinterpreted as a variation of common pregnancy-associated complaints. For the pregnant patient with PAH, symptoms include progressive exertional dyspnea, fatigue, syncope, near syncope, and chest pain, and these symptoms are observed with similar frequency in patients with or without a prior diagnosis.14 Worsening symptoms tend to occur during gestational weeks 20 through 32 as a result of hemodynamic changes brought on by an expanding plasma volume and increased cardiac output.1 Accompanying physical findings include a parasternal heave, cardiac murmur of tricuspid insufficiency and/or congenital heart murmur, jugular venous distention in the upright position, and increasing lower extremity edema. Notably, the symptoms and signs of PAH become more prominent as pregnancy progresses, and patients with a poorer pre-pregnancy functional class (III or IV) will develop symptoms earlier in pregnancy and are at greater risk of hemodynamic collapse (Table 11-2). A systematic review found that 64% of patients were diagnosed with PAH before pregnancy and their functional class ranged from II to IV. In addition, 47% of patients diagnosed before pregnancy were noted to have congenital heart disease-associated PAH and 27% idiopathic PAH.5 In contrast, patients diagnosed during pregnancy were more commonly diagnosed with idiopathic PAH.
World Health Organization (WHO) Functional Classification for Pulmonary Hypertension |

DIAGNOSIS
Transthoracic echocardiography is an essential screening test for pulmonary hypertension. Echocardiography allows noninvasive assessment of cardiac function and provides important information concerning the etiology of pulmonary hypertension.12 This modality allows assessment of right and left ventricular dimensions and function as well as left atrial enlargement and the presence of mitral or aortic valve abnormalities that are associated with pulmonary venous hypertension. In addition, congenital cardiac anomalies such as atrial or ventricular septal defects and patent ductus arteriosus may be identified. Estimation of the pulmonary artery pressure or right ventricular systolic pressure (RVSP) is based on Doppler measurement of the tricuspid regurgitant jet and has been reported to correlate well with measurements of pulmonary artery pressure.15–17 However, in patients with pulmonary hypertension, various investigators have reported over- and underestimation of echo-derived Doppler measurement of the pulmonary artery pressure when compared with right heart catheterization, thereby raising concern about the use of echocardiography to accurately establish the diagnosis of PAH.18,19
The hemodynamic profile as measured by right heart catheterization during pregnancy in PAH patients has been reported in three recent case series. Patients demonstrated a range of elevated right atrial and mean pulmonary artery pressures and relatively low cardiac outputs, 6 to 11 mmHg, 43 to 49 mmHg, and 4.6 to 5.7 L/min, respectively.20–22 In pregnant patients, simultaneous assessment of echocardiography-derived RVSP and right heart catheter measurements of pulmonary artery have demonstrated a good correlation.23,24 However, a study in 18 pregnant patients compared pulmonary artery pressure measurements obtained by echocardiography and right heart catheterization and found that echocardiography overestimated pulmonary artery pressures in one-third of patients.24 Similarly, a retrospective hemodynamic assessment comparing echocardiography and right heart catheterization in 27 pregnant patients reported echocardiographic overestimation in 20 patients.25 Importantly, 32% of patients were found to have normal pulmonary artery pressure measurements as measured by right heart catheterization. Thus, echocardiography is a useful, noninvasive, screening tool to evaluate a patient for the presence or absence of a cardiac condition as the source of their symptoms. However, right heart catheterization is necessary to establish a diagnosis of PAH.12 Because, the diagnosis carries grave implications, it is necessary to confirm the diagnosis by right heart catheterization, especially in a pregnant patient without a prior diagnosis. Moreover, right heart catheterization allows the clinician to distinguish PAH from other causes pulmonary hypertension, such as left ventricular dysfunction or valvular heart disease, and thereby initiate medical therapies specifically developed to treat PAH.
MANAGEMENT
PAH is a significant risk factor for maternal morbidity and mortality; therefore, early termination is recommended by many experts. Counseling is an essential component of the process of care and includes discussion about the effects of medications on the fetus and complications of pregnancy, including maternal death. Parturients with PAH should be managed at a high-risk obstetric hospital by a multidisciplinary group consisting of a high-risk obstetrician, pulmonary hypertension physician specialist, anesthesiologist, neonatologist, and intensive care specialist.5,14
Advances in medical management of PAH have led to improved patient outcomes and this experience has been translated to the care of the pregnant patient with PAH. In 1964, McCaffrey and Dunn described the outcomes of 16 published case reports of pulmonary hypertension in pregnancy and reported a 56% mortality rate.26 Other case series described an unpredictable and complicated pregnancy punctuated by episodes of sudden hemodynamic instability associated with maternal mortality rates of 30% to 50%.27–30 Complications such as hemodynamic collapse from right ventricular failure were observed before and during delivery as well as several weeks postpartum. These reports described poor maternal and fetal outcomes before the commercial availability of PAH-specific therapies; namely, prostacyclin analogues, nitric oxide, and phosphodiesterase inhibitors. Subsequent single center reports described successful maternal and fetal outcomes with fewer complications associated with calcium channel antagonists, prostacyclin analogues, and nitric oxide.30–35 A recent meta-analysis compared the outcomes of published cases concerning pregnancy and PAH managed before (1978-1996) and after (1997-2007) commercial availability of PAH therapies. The authors examined characteristics, management, and outcomes of 73 patients with PAH and reported a decline in maternal mortality from 38% to 25% when comparing the former to the latter era.5 Kiely et al. described their single center experience over 7 years providing care during nine consecutive pregnancies that included inhaled iloprost and reported an 11% mortality.21 Similarly, Jaïs et al. reported the experience concerning management with PAH-specific therapies in 18 patients treated at 13 pulmonary hypertension centers across Europe, Australia, and the United States.20 The authors described a 12% maternal mortality with three maternal deaths and a patient that required urgent heart-lung transplantation. Finally, a study regarding management of 18 patients using PAH medications at five US referral centers found a 16.7% maternal mortality with one hospital death and another death 3 months postpartum.22 Collectively, the observed improvements in maternal outcomes are related to (1) earlier recognition of PAH with echocardiography followed by referral to high-risk obstetrics centers, (2) early prepartum initiation of PAH-specific therapy with continued administration during and after delivery, (3) greater clinical experience in treatment of right ventricular failure associated with PAH, and (4) advances in obstetric medicine and anesthesiology.
FLUID MANAGEMENT
During pregnancy, hemodynamic changes are observed as early as 5 to 8 weeks of gestation and extend several weeks into the postpartum period.36 Notably, increases in plasma volume, left ventricular end-diastolic volume, and stroke volume begin at week 10 and by the third trimester, maternal cardiac output and plasma volume have increased by 30% to 50% compared with pregestational levels.37 Attention to increases in plasma volume and addressing this through implementation of 1.5 to 2 L/day fluid restriction and diuresis has been found to be beneficial.22,38 Reduction in intravascular volume with the use of intravenous diuretics to promote a net negative fluid balance over a 24-hour period with close monitoring of renal function and blood pressure is important.39 In particular, minimizing peripartum and postpartum fluid administration appear to prevent right ventricular compromise. This is highlighted by a case report involving a patient with moderate mitral stenosis and pulmonary hypertension that developed worsening right ventricular function in the postpartum period and right ventricular function improved with the use of oral diuretic therapy.40 In addition, intravenous fluid should be administered with caution as patients with chronic right ventricular dysfunction, as with PAH, may experience hypotension.41 Volume loading in the form of packed red cells or normal saline may produce right ventricular over distention with a paradoxical reduction in left ventricular filling manifested as hypotension.41 The latter is secondary to a shift of the interventricular septum to the left limiting the filling capacity of the left ventricle with a subsequent decrease in cardiac output. Therefore, volume status requires bedside clinical assessment and if uncertainty remains, then the use of echocardiography, measurement of central venous oxygen saturation or placement of pulmonary artery catheter may help determine intravascular volume status.42,43
PULMONARY ARTERIAL HYPERTENSION THERAPY
Compared with conventional therapies, calcium channel antagonists and PAH targeted therapies have been reported to improve exercise capacity, reduce symptoms, and improve survival in nonpregnant patients with PAH. Advances in the diagnosis and medical management of patients with PAH have led to improvements in symptoms, exercise tolerance, and survival.12 Table 11-3 shows the FDA category of the most common used agents. Historically, calcium channel antagonists were the first medical therapy demonstrated to have clinical efficacy for the treatment of idiopathic PAH.44 Before initiation of a calcium channel antagonist, placement of a pulmonary artery catheter and concomitant administration of inhaled nitric oxide or intravenous epoprostenol is required to identify patients that will exhibit pulmonary vascular vasoreactivity.7,12 Once a favorable hemodynamic profile is identified with the use of the short-acting agent, the calcium channel antagonists is cautiously started while using a pulmonary artery catheter to measure and record hemodynamics. A retrospective study of 557 patients demonstrated that 70 patients (12.7%) demonstrated acute vasoreactivity defined as a reduction in mean pulmonary artery pressure and pulmonary vascular resistance ≥20 % of baseline.45 Moreover, a vasoreactive response was principally observed in patients with idiopathic and heritable PAH and those patients demonstrating acute vasoreactivity, no more than half demonstrated long-term benefit from calcium channel antagonists. Thus, only a minority of patients will have lasting hemodynamic improvements with calcium channel antagonists and they should only be used by clinicians experienced with acute vasodilator testing.45
FDA Safety Ratings of Medications in Pulmonary Arterial Hypertension Treatment |
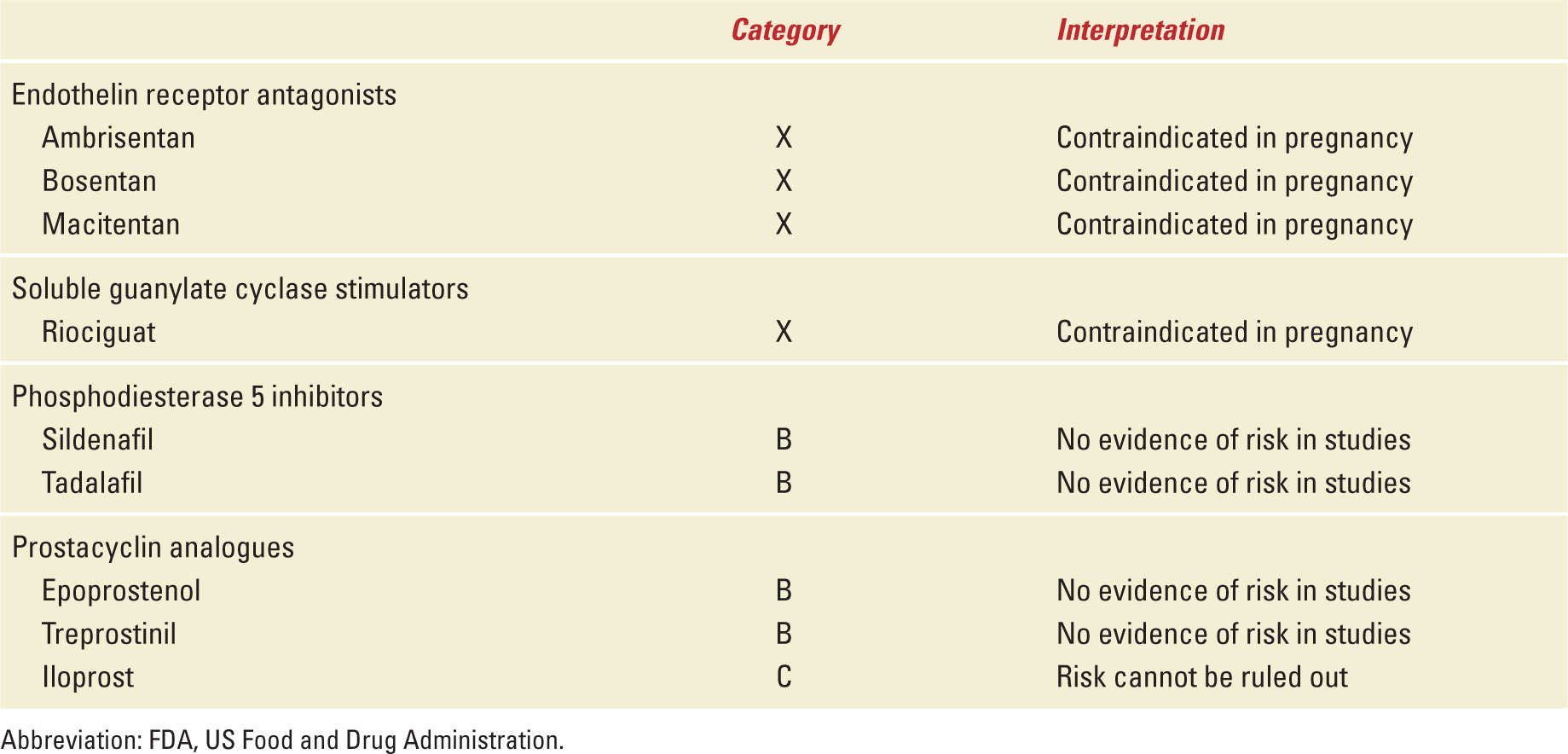
Over the past two decades, four classes of medications targeting the pulmonary vascular endothelium have been developed and there are currently nine US Food and Drug Administration approved medical therapies for treatment of PAH (Table 11-4). Prostacyclin analogues are administered to increase low levels of prostacyclin produced by decreased expression of endothelial prostacyclin synthase. Compounds targeting the prostacyclin pathway include parenteral, subcutaneous, inhaled, and oral prostacyclin analogues.46 Chronic administration of prostacyclin analogues have been used to treat right ventricular dysfunction related to PAH.46,47 Administration of epoprostenol (Flolan, GlaxoSmithKline, Philadelphia, PA) has been demonstrated to improve hemodynamic profile in parturients with idiopathic PAH and PAH associated with scleroderma, HIV, congenital heart disease, portal hypertension, and drugs.5,33,48 In addition, case series have described use of intravenous epoprostenol or treprostinil (Remodulin, United Therapeutics, Research Triangle, NC) in parturients with PAH with good outcomes and no reported congenital anomalies.20,22 Subcutaneous treprostinil has been used during pregnancy and associated with a maternal death.20
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


