Fig. 19.1
Reciprocal errors and PSSC. Impact of the type of meiotic error and the predictive value of polar body (PB) screening results and the eventual chromosomal composition of the embryo. Note that reciprocal errors prognosticate aneuploidy after non-disjunction but euploidy after premature separation of sister chromatids (PSSC) making that result indeterminate. Adapted from [90]

Fig. 19.2
PSSC with correction. Data from an oocyte that underwent first and second polar body analysis and subsequent embryo biopsy. The polar bodies demonstrated reciprocal errors. Given that the embryo was normal, this represents premature separation of sister chromatids with correction of the meiosis I error in meiosis II. The reproductive competence of the embryo was demonstrated by the delivery of a healthy infant. Adapted from [90]
19.3.2 Blastomere Biopsy
Biopsy of the embryo itself is essential to attain the greatest opportunity to capture aneuploidy. This may be done at the cleavage stage prior to compaction or later in development after blastocyst formation. Removal of cells from two- and four-cell embryos is known to reduce the inner cell mass (ICM), the portion of the embryo from which the fetus is derived [68]. Common practice has been to biopsy embryos at the eight cell stage on the morning of day 3. Indeed, until recently 88% of biopsies for preimplantation genetic diagnosis (PGD) cases were performed on day 3 cleavage stage embryos, with less than 1% being performed at the later blastocyst stage [55]. Greater concerns arise when more than one cell is required, as is the case with many platforms capable of PGD.
The safety of biopsy at the eight cell stage was based upon the fact that all cells at this stage are thought to be totipotent [69] and that removal of cells did not appear to compromise ICM allocation [70]. However, both the cells and cytoplasm have become polarized at this stage and given that all cells are morphologically the same, it is not possible to determine which cells are destined to become fetal and may be more likely to be detrimental to development if biopsied. Finally, the number of cells available at the cleavage stage limits the number of cells that can safely be removed and thus limits the starting material available for amplification and analysis . Removal of even one blastomere affects outcomes [18] and two blastomeres rather than one blastomere has been shown to be detrimental to ultimate blastocyst formation [71].
19.3.3 Trophectoderm Biopsy
The final stage of development prior to embryo transfer during IVF is the blastocyst stage. At this point the blastocoelic cavity has begun to develop and the trophectoderm cells, those destined to be amniotic membranes, and the ICM cells, those destined to become the embryo, can be easily distinguished. It is possible to remove up to five or six cells, which can be used to reliably reduce no result rates due to failed amplification when using platforms for CCS. Initially, trophectoderm biopsy was utilized as confirmatory diagnosis following a polar body or blastomere biopsy [72].
Until only recently, no class I data existed evaluating the safety of embryo biopsy at these two developmental stages: cleavage stage and blastocyst biopsy of trophectoderm. The major limitation of literature comparing the techniques was selection of controls. Given the large variety of reasons patients seek preimplantation genetic diagnosis, it can be challenging to compare two groups of patients. Recently, a blinded, paired, randomized trial was undertaken to determine the impact of embryonic biopsy [73]. This experiment was done for both cleavage stage biopsies and for trophectoderm biopsy at the blastocyst stage and showed a relative reduction of 30% in the sustained implantation rate for blastomere biopsy.
There are several possible reasons for these results. First, the trophectoderm biopsy removes proportionally fewer cells than the cleavage stage biopsy [74]. One or two blastomeres taken from an eight cell cleavage stage embryo represents 12.5–25% of the embryo, whereas four to five trophectoderm cells taken from an expanded blastocyst containing 200–220 cells represent only 2–3% of the total cells. Another possibility is that, as noted above, trophectoderm cells represent an extraembryonic component of the blastocyst whereas the specific fate of the blastomere being removed cannot be determined by morphology, and thus could represent cells with gene activation that have destined them to become the embryo [75]. The final possibility is that blastocysts simply tolerate mechanical manipulation better than cleavage stage embryos [76].
Thus, trophectoderm biopsy of four to five cells from an expanded blastocyst represents the safest way to obtain cells from a developing embryo that contains the full maternal and paternal complement of DNA that will become the conceptus. In light of the highly predictive nature of the chromosome complement in the trophectoderm for the inner cell mass, blastomere biopsy represents an unnecessary risk [77].
Embryonic mosaicism is important to note here. It is known that FISH overestimates true-cleavage stage mosaicism [78] in some cases indicating it occurs as much as 100% of the time. There is data utilizing SNP microarray which suggests it occurs 24–31% of the time [41, 78]. These mosaicism rates may account for the failures of euploid blastocyst transfers. Additionally, mosaicism likely accounts for the majority of misdiagnoses made when utilizing a validated PGD platform [79].
19.4 Strategies for Single Gene Defect PGD
There were a number of limits which have faced PGD in its infancy. They primarily relate to the fidelity of amplification and analysis of samples with extremely limited amounts of starting material. This was overcome by analyzing the DNA in multiple areas, primarily through linkage analysis. This was done by finding single nucleotide polymorphisms (SNPs) on the impacted chromosome which are different than those on the normal chromosome for each individual. Then, at the time of analysis, multiplex PCR reactions are utilized to look not only for the mutation but also for the linked markers which should be nearby. In doing this, the laboratory has several chances to detect the presence or absence of the mutation.
Unfortunately, linkage analysis is not always straightforward. The following are some of the factors that may impact the reliability of this approach:
- 1.
It may not be possible to target the mutation directly. In such cases, additional linked markers are recommended
- 2.
The potential for recombination exists. Recombination occurs during meiosis I and at that time DNA is exchanged between chromosomes. If the point of recombination lies between the marker and the mutation, then the marker will be moved to the normal chromosome. In this setting, the assay might perform perfectly and still prognosticate the wrong clinical result! There are ways to minimize the risk that recombination will impact the validity of the screening result [55]:
- (a)
Pick markers which are as close to the mutation as possible. The closer the mutation the less likely it is that a recombination event will occur between them.
- (b)
Ideally the marker would be intragenic and exhibit a high degree of heterozygosity and provide consistent high quality amplification
- (c)
Extragenic markers which are utilized should ideally be within 1 MB of the mutation
- (d)
Use a total of four markers—two upstream and two downstream of the mutation. Thus if ADO occurs but there is concurrence with the other three a result may still be determined. If only a single marker is used—specifically in those cases where the mutation is not being evaluated directly—an indeterminate will result for any discrepancy
- (e)
Recombination will be reflected by a discrepancy between the more proximal and distal markers. This phenomenon should be rare (~1%). If you have several embryos in a cohort with recombination in the interpreted results, it may be prudent to reevaluate the screening strategy or the fidelity of the amplification for those markers
- (a)
19.4.1 Alternative Strategies for Single Gene Defect PGD
The wide spread availability of cost effective NextGen sequencing platforms may impact single gene defect PGD. A recent has demonstrated the ability to sequence through the area of the defect providing both qualitative and quantitative information [49]. This would functionally provide more markers and the depth of the sequencing appears to meaningfully enhance the precision of these assays. With the appropriate amplification strategies, it may also allow simultaneous aneuploidy screening which would empower the transfer of lower numbers of embryos while lowering loss rates and improving outcomes. There is much work to be done as the validations are not yet published.
19.5 Strategies for Aneuploidy PGS
Amplification is a critical step in the molecular analysis of these samples. There are approximately 6 pg of DNA in a single cell. Thus, an average trophectoderm biopsy of five cells would provide approximately 30 pg of DNA. Technologies capable of copy number analyses require dramatically more DNA to obtain a result: typically several hundred nanograms or more. This requires a 100,000 (or more) fold amplification of the DNA in a relatively uniform fashion.
Many techniques employ whole genome amplification. While it has the advantage of potentially amplifying the entire genome, uniformity is typically not attained and may vary as much as several thousand fold across different portions of the genome. This disadvantage may be overcome by evaluating the chromosomes at a large number of different sites and then performing statistical smoothing. Alternatively, a targeted approach focused at specific loci on each chromosome may be used. This is more uniform but still varies and requires some degree of statistical smoothing. Whole genome amplification is required for SNP microarray and CGH related technologies. In contrast, qPCR is done with targeted amplification of specific loci and next generation sequencing (NGS) may be done with either.
One metric by which to compare the available technologies is the time required to perform the test. This is of great importance in the application to real-time IVF. Often, the embryo is transferred in the same cycle as the oocyte was retrieved, a so-called fresh embryo transfer. If this is not possible, the embryo must be subjected to freezing and transferred at a later day, a so-called frozen transfer. As discussed, the biopsy is most optimally performed on the expanded blastocyst. This leaves less than 24 h within which to make a diagnosis and transfer the embryo in that fresh cycle. If diagnosis takes longer than this, embryos must be frozen, using either slow freezing methods or vitrification, and then thawed after the diagnosis is made and transferred in a separate cycle. In circumstances where results cannot be obtained in time for a fresh embryo transfer, modern vitrification techniques yield equivalent pregnancy outcomes in fresh or frozen cycles [10].
Traditional metaphase CGH requires a minimum of 72 h to yield results. There are shorter mCGH protocols, but even these take up to 24 h for the entire process to be completed and results may not available in time for a fresh transfer [59, 60, 80–82]. Thus, aCGH is an attractive alternative, requiring only 12 h for results [83], with the added benefit of greater throughput. SNP arrays have also been used to perform CCS on blastomeres in time for a fresh transfer and also have a result rate time of 12 h [46]. Finally, qPCR results are available in as little as 4 h which enables a fresh embryo transfer after analysis of trophectoderm cells from an expanded blastocyst [84]. Thus, only qPCR analysis allows for safe biopsy of a day 5 blastocyst with results in time for a fresh transfer early on day 6.
Thus, given the ability to have results relatively quickly, DNA array technology was widely applied to CCS in the form of aCGH and SNP array. There are two main contrasting attributes of these two platforms. First is the number of DNA base probes across the 24 chromosomes. Although accuracy cannot be precisely correlated with probe number, resolution, which in CCS is the ability to distinguish between euploid and aneuploid, varies significantly. For example, BlueGnome (Illumina, USA), a widely used aCGH platform uses bacterial artificial chromosome (BAC) arrays, and possesses between 2000 and 5000 DNA probes spread across the 24 chromosomes [85]. By comparison, the Agilent (Agilent, USA) utilizes 180,000 oligonucleotide probes [48] and the Affymetrix (USA) SNP array contains 262,000–370,000 probes [41, 46, 47].
In addition to resolution differences, the way that copy number is assigned for each chromosome differs between aCGH and SNP arrays. With aCGH copy number is assigned by analyzing a ratio of red (sample) and green (control) fluorescence after differential labeling and mixing of biopsy DNA with control DNA [86]. With the Affymetrix SNP array, a single color system is used where the fluorophore hybridizes only to the biopsied DNA sample and then computational comparisons of signal intensities are made to separate control DNA hybridized arrays [87]. The former has the advantage of comparing a control sample in the same time frame under the same laboratory conditions. The latter has the advantage of comparing the sample DNA to several control samples, rather than just one, which allows for better statistical smoothing of natural inconsistencies in control samples. Furthermore, the differences in the paradigm utilized to assign copy number impact the platform’s ability to gather further information from the test. For example, SNP arrays allow for a determination of parental origin of both monosomy and trisomy of parental origin from the SNP array data alone [67]. Additionally, uniparental isodisomy, although a rare event, has been validated utilizing SNP array through loss of heterozygosity analysis [5]. Finally, DNA fingerprinting from SNP array data provides clinicians with the opportunity to prevent misdiagnosis from contamination and can also be used to track which embryos implant in the event of a multiple embryo transfer for research and clinical purposes screening [88]. The array CGH platform is unable to provide these additional points of information.
19.5.1 Clinical Impact
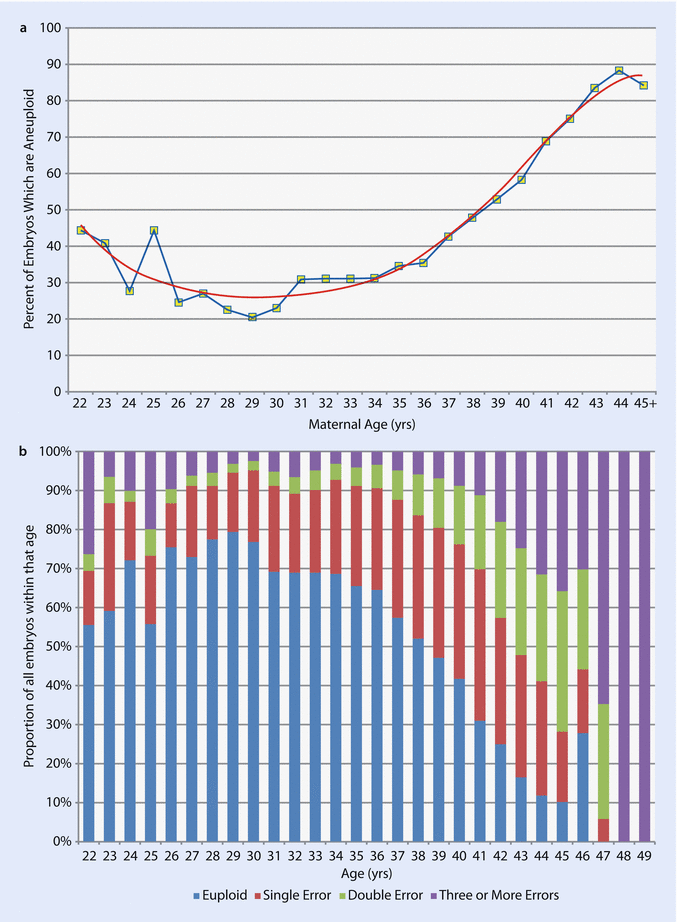
Intuitively it seems logical that PGS would improve outcome with older patients. Aneuploidy rate is closely related to maternal age (◘ Fig. 19.3a). Furthermore the complexity of the aneuploidy increases with age (◘ Fig. 19.3b). The fact that a platform is accurate and reliable and has known positive predictive value does not indicate that clinical outcomes can be improved by applying the technology. This is the final step in validation: a prospective, randomized selection study. This class I data was recently obtained and demonstrated that CCS for aneuploidy utilizing qPCR, which had been cross-validated with SNP array, meaningfully enhances embryo selection and ultimately implantation and delivery rates [90]. Indeed, in this study, sustained implantation rates in the trophectoderm biopsy and CCS group was 66.4% versus 47.9% in the control group. Subsequent class one data proved the single embryo transfer of a CCS screened blastocyst had equivalent outcomes as double embryo transfer in an unscreened arm with elimination of twin pregnancies [10]. Unfortunately, not all the platforms in use for CCS have undergone rigorous validation. However, many of the shortcomings have been addressed and continued work will hopefully alleviate this concern. It is critical that this occurs lest their use results in a similar story to that of FISH.