10. Pharmacology in Neonatal Care*
Mary Miller-Bell, C. Michael Cotton and Deanne Buschbach
Optimal pharmacotherapy delivers the maximum intended beneficial effect with the minimum toxicity. Determining the optimal pharmacotherapy for neonates is problematic in that much of the data have been extrapolated from research in adults, children, and laboratory animals. Neonates show dramatic differences in the way they respond to drugs compared with older children and adults and within the neonatal population. 18 Gestational age, chronologic age, and disease state alter a neonate’s ability to metabolize medications and affect the infant’s response to the drug.
This chapter discusses pharmacology as it relates to the neonate and illustrates how rational medication decisions can be made for neonatal intensive care unit (NICU) patients. It also discusses strategies to avoid medication errors, strategies for drug delivery, and the new frontier of a genetic variation–based approach to pharmacotherapy.
PHYSIOLOGY
Pharmacodynamics and Pharmacokinetics
The drug-receptor theory states that the amount and duration of a drug’s availability to a receptor determine its effectiveness. Pharmacodynamics describes what the drug does to the body, whereas pharmacokinetics describes what the body does to the drug, how much drug is available to the receptors, and for how long (Figure 10-1). 12 A drug’s disposition can be described by four processes: drug entry (absorption), distribution, biotransformation, and elimination.
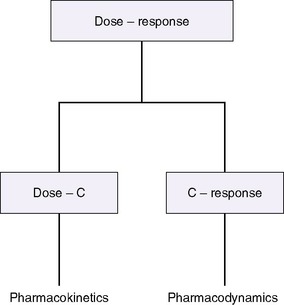
Pharmacodynamics relates the amount of available drug (or active metabolite) to the effect and depends on receptor availability, affinity of the drug for the receptor, and cellular function.
Antagonist drugs block a receptor’s cellular and physiologic activity (e.g., naloxone), whereas agonist drugs elicit the receptor’s action (e.g., cardiovascular agents such as dopamine and epinephrine). Some drugs act with receptors to increase or decrease gene expression (e.g., antenatal steroids), whereas others affect cell membrane permeability. Some drugs, such as methylxanthines, increase or decrease the amount or activity of “second messenger” molecules within cells. Antibiotics and antiviral agents act through some of these mechanisms to reduce the viability of pathogenic organisms by changing vital characteristics and functions. Readers should note that most drugs have more than one effect, so although the desired therapeutic effect may occur, the drug’s other effects can limit its usefulness. Side effects, which can vary from the minor to the prohibitive, occur within the therapeutic range of concentration. Toxic effects result from drug overdose or serum concentrations higher than the recommended therapeutic range.
Individual infants may have idiosyncratic responses to medications, as well as expected responses. Infants who are low sensitivity responders exhibit a drug response less than that expected for a usual dose, whereas infants who are extreme sensitivity responders exceed the expected response for a given dose and drug level. Unpredictable adverse reactions differ from expected responses. Patients may become tolerant to a given drug dosage, as is commonly seen with opiates. Tachyphylaxis, a rapid decrease in drug response without a dosage change, may be related to limited receptors or other intracellular mechanisms. 3,10
Developmental differences in number and function of receptors and intracellular mechanisms are critical to estimating drug actions. An example of developmental effect on pharmacodynamics is the diminished sensitivity of the cardiovascular system to digitalis in the youngest patients; the receptor number increases with age. Changes in alpha- and beta-adrenergic receptors also occur with gestational and chronologic age and must be considered in determining dosage with pressors and inotropes. 12
To elicit the desired therapeutic effect, the drug must be delivered to the receptor and remain available for an appropriate amount of time. 29
Pharmacokinetics describes the delivery and removal of the drug to and from the body. Doses and dose intervals are expressed mathematically by pharmacokinetic disposition parameters related to distribution, biotransformation, and elimination, such as clearance, volume of distribution, and half-life.
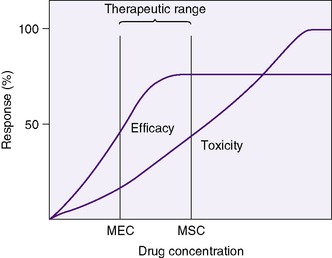
A clinician bases drug choice and dose regimen largely on the desired therapeutic response and toxic effect in “average” patients. Plasma concentration provides a surrogate for effect when the relationship between concentration (C) and effect has been demonstrated in similar patients. The minimum effective concentration (MEC) is that at which 50% of patients exhibit the desired response (Box 10-1). The maximum safe concentration (MSC) is that at which 50% of patients exhibit a toxic response (Figure 10-2). To continue the desired effect, the clinician aims to obtain a target plasma concentration at “steady state” (Css), somewhere between the MEC and the MSC, where most patients exhibit the desired effect and few suffer toxic effects. With ideal maintenance therapy, drug input equals drug elimination. The variability around the Css depends on dose, dose interval, and drug disposition.
BOX 10-1
| C | Drug concentration (plasma or serum) | mg/L |
| Css | Steady state concentration (average) | mg/L |
| MEC | Minimum effective concentration | mg/L |
| MSC | Maximum safe concentration | mg/L |
| F | Extent of drug availability (0-1): how much active drug gets to the systemic circulation | Unitless |
| Vd | Volume of distribution: relates to loading dose | L/kg |
| Cl | Clearance: relates to maintenance dose | L/kg/hr |
| t ½ | Drug elimination half-life: relates to the time course of changes in drug concentration | Hours |
L, Liter = 1000 milliliters; mg, milligram = 1000 micrograms.
The target Css is influenced by the amount of drug bound to plasma protein. In a newborn, free unconjugated bilirubin can displace numerous medicines of lower protein affinity, and numerous medicines can displace unconjugated bilirubin, increasing unconjugated bilirubin’s serum concentration and its potential for toxicity. Intravenous (IV) lipid infusions also can affect protein binding of both bilirubin and some medicines. The drug concentration measured in most available assays is usually the total, both protein-bound and free; therefore the available concentration at the receptor usually is somewhat less than the total serum concentration.
DOSE-CONCENTRATION CONSIDERATIONS RELATED TO AGE
The reported therapeutic range for theophylline in adults is 10 to 20 mcg/mL for bronchodilation. In neonates, the drug has been used to treat apnea of prematurity, and the effective range for this disorder has been 4 to 12 mcg/mL. 29 Theophylline is reported to be 36% bound to plasma protein at a total concentration of 8 mcg/mL in newborns, compared with 70% bound in adults. A total theophylline concentration of 10 mcg/mL in an adult represents 3 mcg/mL free theophylline available to receptors, whereas in a neonate, a total of 4.7 mcg/mL equals 3 mcg/mL of free theophylline (also, the metabolism of theophylline in neonates leads to measurable free caffeine). Decreased bound theophylline in neonates could explain why therapeutic effect is achieved with lower total serum concentration in neonates.
In the NICU, doses and intervals must be adjusted based on changes in the dose-concentration and the concentration-response relationships. This enables a more accurate and precise response to changes in dose-concentration effects, especially total concentrations, but as with the theophylline case, we must watch for clinical effects and estimate other factors’ influences to estimate the free concentration’s effectiveness and the receptor and cellular responsiveness. Potential causes of changes in dose-concentration relationships unique to newborns are described for the pharmacokinetic processes that follow.
Absorption
The process of absorption defines the rate and amount of drug that enters the bloodstream. The parameter F indicates the percentage of dispensed drug available in the systemic circulation, with F = 1 indicating the drug is 100% available. We lack systematic studies of absorption in sick newborns, and differences in absorptive processes are expected but remain unmeasured generally. Some differences in newborns that potentially affect bioavailability include developmental changes in surface area and permeability of gastrointestinal (GI) mucosa, age-dependent changes in acid secretion in the stomach (higher pH than in older children and adults), changes in gastric emptying time and total GI transit time, and the characteristics of GI flora. Drugs such as ranitidine and metoclopramide also affect absorption of other medications by means of the same mechanisms.
“First-pass” pharmacokinetics means the drug is absorbed through the GI mucosa and travels directly to the liver, where it is metabolized and excreted in significant amounts, limiting bioavailability. Different drugs are absorbed at different rates, and different formulations of the same medication may be protected from first-pass metabolism. Drugs also may be given by inhalation, intranasally, intrarectally, topically, intramuscularly, subcutaneously, and intravenously. 12
Distribution
Medications rely on blood flow and drug solubility for distribution to their sites of therapeutic effect. The volume of distribution for a drug is a parameter that relates total amount of drug distributed throughout the body to the serum or plasma concentration.3 It is an attempt to quantify the space in which the drug can go. Strictly defined, it is the hypothetical volume of body fluid necessary to dissolve the total amount of drug as found in the serum. Volume of distribution must be used to estimate the amount of a loading dose or a change in plasma concentration with any bolus dose:
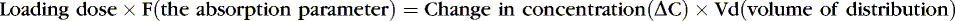
Or, put another way:
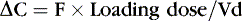
Volume of distribution usually is expressed as a function of body weight, with units of volume per kilogram. Major factors that affect distribution volume are plasma protein binding and body composition. 12,18,29 Changes in body composition happen throughout fetal and newborn life. Total body water decreases with increasing age: 85% in the smallest, most preterm infants; 70% in term infants; and 55% in most adults. Total body water may increase with such conditions as the syndrome of inappropriate antidiuretic hormone (SIADH) excretion, which increases total body water. Extracellular water composes about half this amount in a healthy term neonate. Large water-soluble molecules reach this compartment. Intravascular water composes about 10% of the body weight; protein-bound medications stay in this small compartment. Water-soluble drugs such as penicillins, aminoglycosides, and cephalosporins are distributed in a greater volume in smaller, more preterm infants, therefore requiring a higher loading dose per kilogram, if total body water were the only determinant of volume of distribution.
Plasma protein amounts and binding capacities also differ with gestational and chronologic age. Protein binding is decreased in newborns, because lower amounts of albumin are available than later in life, and fetal albumin has less capacity to bind some drugs. Acidic drugs such as ampicillin, phenytoin, and phenobarbital bind less well, thus increasing the free (available to receptor) fraction of the drug, with resultant increase in effect. Changes in pH also can affect a drug’s affinity for albumin. Fat content varies with gestational age and degree of illness; increased fat content increases the volume of distribution. Lipid-soluble molecules also are distributed in this space.
Of particular concern in newborns is the interaction of circulating unconjugated bilirubin and protein-bound drugs. Several anionic compounds bind to albumin and can displace bilirubin, increasing free bilirubin, thus increasing its potential for toxicity. Bilirubin has a higher affinity for albumin than some other medications; it may displace them from albumin, increasing the medication’s availability and potential to reach toxic levels.
Biotransformation
Biotransformation, or drug metabolism, occurs most commonly in the liver. Phase I metabolism describes the nonsynthetic metabolism of medications. Phase II, usually conjugation, or the addition of a substance to a medication, is synthetic metabolism. Oxidation, conjugation, glucuronidation, and hepatic blood flow change with gestational and chronologic age, diseased states, and use of certain medications. For example, oxidation and glucuronidation are decreased in newborns. Drugs such as acetaminophen, phenobarbital, and phenytoin, which require oxidation for elimination, remain available longer and may be transformed to other active metabolites (the neonatal liver metabolizes theophylline to caffeine), or the drug may remain at significant free concentrations for a prolonged period. The possibility of prolonged peak concentrations of available drug or active metabolites for many pharmaceuticals mandates careful monitoring of drug levels and clinical conditions to titrate dose intervals. To further the potential for confusion and trouble and further the argument for careful assessment of levels and clinical signs of effectiveness and toxicity, a decrease in plasma protein binding (or any other change in volume of distribution) may increase the hepatic clearance of a drug.
Clearance (Elimination)
Drug clearance or elimination occurs by excretion of unaltered drug or biotransformation to an inactive metabolite. Most drug elimination pathways can become saturated if the dose is high enough and dose intervals are too frequent. Most drugs in use in the NICU have therapeutic doses less than those necessary to saturate the elimination system. When clearance mechanisms are not saturated, the Css in plasma is proportional to the dose rate. Clearance equals the rate of drug elimination divided by the drug concentration. 19 Just as volume of distribution relates to loading dose and initial concentration, clearance relates to a maintenance dose that keeps a drug’s concentration at steady state. So for an ideal drug maintained at steady-state concentration:
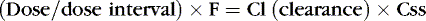
Stated another way:
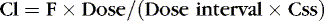
Or, to tailor the dose for a desired steady-state concentration:
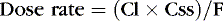
The appropriate dosing rate can be calculated if the clinician can specify the desired steady-state plasma concentration and knows the clearance and bioavailability of a drug (from peak and trough levels in a particular patient).
For example, clearance of theophylline in preterm infants is reported to be 0.017 L/kg/hr. If the desired Css = 8 mg/L, assuming F = 1, particularly if the dose is to be given intravenously:
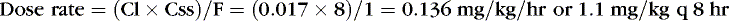
RENAL EXCRETION
The kidney is the primary route of excretion for many drugs commonly used in the NICU. The kidney clears drugs through glomerular filtration and tubular secretion. Examples of medications eliminated through the kidney are aminoglycosides, digoxin, diuretics, and penicillins. Doses and dose intervals of drugs that have renal excretion must change with age and disease state. The glomerular filtration rate (GFR) (i.e., the amount of blood filtered by the kidney in a unit of time) is low at birth and gradually increases over the first weeks. In preterm infants, the GFR starts even lower than in term infants, with a somewhat significant increase occurring at 34 weeks after conception. Tubular secretion also matures with increasing gestational age and depends on tubular function. In adults, aminoglycosides may be dispensed based on creatinine clearance, but in neonates less than 1 week old, serum creatinine may reflect maternal levels, as well as renal impairment. Acidosis and a history of hypoxia or ischemia also may modify an infant’s renal function, slowing excretion and altering pharmacokinetics. Again, measuring levels in cases of suspected renal impairment, whether from suspicious history or laboratory values, is important to determining an appropriate dosing strategy.
Half-Life
A drug’s “half-life” (t ½) is the time necessary for the drug level to decline by 50%. Half-life is related to both volume of distribution (Vd) and clearance (Cl), so that:
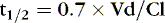
The t ½ is used to predict and interpret the time course of changes in plasma drug concentrations. For example, the time to steady state is 4 to 5 half-lives. The half-life is useful in selecting dose intervals. This concept is illustrated in Figure 10-3.
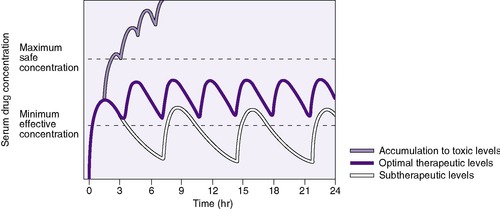 |
| FIGURE 10-3 (From Roberts RJ: Drug therapy in infants, Philadelphia, 1984, Saunders.) |
Loading doses help expedite reaching desired therapeutic concentrations, especially for drugs with long half-lives, in which a desired effect is needed immediately. For drugs with one-compartment distribution that stay in the circulation and are not stored in cells or tissue, the loading dose may be given as a simple single dose. Drugs that are fat soluble or stored intracellularly are more difficult to assess, and therapeutic levels must be included in the loading dose assessment.
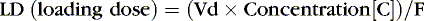
If the volume of distribution for theophylline in preterm infants is 0.7 L/kg, and 8 mg/L is the desired concentration, and F, for an intravenous dose, is assumed to be 1, a loading dose can be calculated.
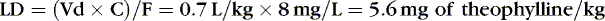
Pharmacogenetics and Pharmacogenomics
In the 30,000-plus known genes, there are over 4 million “common” variants (occurring in more than 1% of the population), many of which directly affect the function of the coded protein. In addition to the multiple factors like age, gender, disease, and concurrent medications, a patient’s genetic code can cause variation in the response to a particular drug. Once a drug is administered, it is absorbed and distributed to its site of action, then interacts with targets, and is finally metabolized and excreted. The enzymes and other compounds involved in each of these processes are subject to genetic variation that leads to variation in function. Pharmacogenetics is the study of the role of inheritance in the individual variation in drug response. Pharmacogenomics is the study of the influence of multiple genes and their interaction with each other and the environment on drug effects. 5,24,28
The first description, over 40 years ago, of a genetically caused variation in drug metabolism was for the enzyme responsible for hydrolysis of succinylcholine. One in 3500 people are homozygous for a gene encoding an atypical form of the enzyme butyrylcholinesterase, which is quite slow to hydrolyze the succinylcholine, leading to prolonged muscle paralysis. Concurrently, an enzyme responsible for N-acetylation, another form of drug metabolism, was found to have a common genetic variant, so some patients are fast metabolizers and others are slow metabolizers of drugs such as hydralazine.
The cytochrome P-450 enzymes are important in phase I drug metabolism. One in particular, CYP-450 2D6, responsible for metabolism of many drugs, has been extensively studied. About 5% to 10% of the adult Caucasian population have genetic variants of the enzyme, leading to decreased activity and higher and more prolonged active drug levels.
Recently, a genetic variant in mitochondrial DNA, the A1555G mutation gene, has been linked to risk for hearing loss associated with aminoglycoside toxicity. The risk-additive variant’s frequency in the general population is estimated between 1% and 3%, but among deaf subjects tested, concurrence of deafness with aminoglycoside treatment with this mutation is quite common. This could lead to future testing before use of aminoglycosides. It is unknown whether tight control of aminoglycoside levels would reduce risk for hearing loss. No recommendations can be made until more extensive, population-based studies are done. Such studies must include accounting for drug levels and duration and genotypes in assessment of risk for hearing loss. 21
In addition to genetic variation in drug metabolism, the genetic polymorphisms of drug targets—including adrenergic and dopamine receptors and enzymes such as acetylcholinesterase—are likely to have effects on the response to drugs targeting these proteins. This may occur with the use of medications such as bronchodilators, pressors, and inotropes, and ACE-inhibitors such as enalapril and captopril.
The study of pharmacogenetics and pharmacogenomics is new, especially to neonatology, in which little is known about pharmacokinetics and pharmacodynamics of commonly used medications. The genetic revolution that has come with completion of the sequencing of the entire human genome is on its way to reaching neonatology with the availability of rapid analysis of large population-based samples for thousands of genes and their variants and how they relate to drug response. In the future, these studies will lead to improved understanding of individual variation in drug response, which should allow development of strategies to individualize care and help avoid complications resulting from heretofore unexplained genetic variations. The challenge will be to understand how the multiple genes and environmental factors interact in individual infants to alter risk for disease and adverse drug responses in fragile infants.
DATA COLLECTION
Clinicians should be aware of a medication’s desired effects, side effects, and toxicities; know when they are expected to occur; and monitor for these effects. Whether a dose effect occurs or not should be noted. Dose–plasma concentration results should be recorded when therapeutic drug monitoring is done. If the drug’s serum concentration relates to clinical response, the blood concentration should be followed in addition to clinical signs. To optimally use drug serum levels, the expected blood concentration is calculated from the dosing history, and patient variables that may affect pharmacokinetics with the timing of blood samples are considered. A comparison of expected values with measured values allows rational adjustment of future dosing. 4 Potential explanations for differences between measured and expected concentrations are listed in Box 10-2.
BOX 10-2




• Inadequate compliance
• Inadequate medication delivery
• Inappropriate timing of samples
• Laboratory error
• Revision in initial estimates of necessary pK required
pK, Pharmacokinetics.
Even if predictable pharmacokinetic and pharmacodynamic changes are considered, other factors may influence a drug’s effect. Clinical end-points must be followed and recorded and dose regimens adjusted accordingly. One example is the monitoring of renal function with indomethacin dosing: If clinical signs of renal dysfunction are noted, the drug is not administered. A pharmacist should be included in the caregiving team to clarify dose and disposition parameters for individual patients with their various conditions.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
