Chapter 14
Pharmacology and Nonanesthetic Drugs During Pregnancy and Lactation
Tony Gin MBChB, MD, FANZCA, FHKAM, Jerome Yankowitz MD
Chapter Outline
Drug therapy during pregnancy can be complex because the physiologic changes of pregnancy may alter drug disposition and effect. Maternal medications may have direct effects on the fetus after placental transfer or indirect effects through changes in placental and uterine function. Even after delivery, drug transfer to breast milk may be a concern. Nevertheless, pregnant women still require medications to treat many acute and chronic conditions. The challenge is finding the balance between the benefits and risks of therapy.
It would seem prudent to use only drugs considered safe in pregnancy. Unfortunately, the potential adverse effects of many drugs remain unclear. Pregnant women are not usually included in early clinical trials, and because of the low incidence of some complications, the first suggestion of adverse effects may be revealed only from post-marketing surveillance and registries of complications. This uncertainty about safety, and the difficulties in determining what public information is reliable, may convince mothers to refuse appropriate drug treatment. New improved drugs are available in many areas of therapeutics, but many prescribers prefer to use older drugs that have a longer empirical history of safety.
Maternal variations in drug effect are not usually difficult to manage, and a change in dosing regimen or the choice of drug may be all that is required. The greatest fears and concerns are for potential fetal effects that may manifest as teratogenicity with fetal loss or congenital malformations, fetal growth restriction (also known as intrauterine growth restriction), preterm labor, and other complications. Impaired development and behavioral problems may manifest after delivery.
The anesthesia provider should understand the implications of pregnancy on drug disposition and effect. Although not usually responsible for primary maternal drug therapy, he or she will encounter women taking many different medications and may sometimes need to administer a variety of nonanesthetic drugs during the peripartum period, either for maintenance of chronic therapy or for acute indications, especially when managing critically ill patients.
In this chapter, how maternal physiology affects pharmacology is summarized, teratology and fetal effects of the main classes of drugs the anesthesia provider is likely to encounter are addressed, and drug transfer to breast milk is reviewed. The more common perioperative drugs are used as examples. Specific drugs used in the management of individual obstetric conditions are discussed in other chapters.
Changes in Drug Disposition and Effect
Pharmacogenetics
Genetic differences are responsible for some of the variation in drug response among individuals. Pregnancy does not obviously modify these pharmacogenetic differences, although some obstetric conditions such as preeclampsia are related to complex genetic factors. There are, however, some examples in which underlying genetic differences do affect obstetric management.1
The metabolism of codeine to morphine is greatly affected by polymorphisms of the cytochrome P450 (CYP) isoenzyme CYP2D6. It has been recognized only recently that mothers who are ultrarapid metabolizers may produce and transfer sufficient morphine through breast milk to cause neonatal central nervous system (CNS) depression and even death.2
Two of the possible changes at the β2-adrenergic receptor are an arginine-to-glycine substitution at codon 16 (Arg16Gly) and a glutamine-to-glutamate substitution at codon 27 (Glu27Gln). When β2-receptor agonists were used for tocolysis, Arg16 homozygotes had longer gestation and better neonatal outcome.3 In the management of hypotension during spinal anesthesia for cesarean delivery, Gly16 homozygotes and Glu27 homozygotes required less ephedrine.4
The µ-opioid receptor gene may have an adenine-to-guanine substitution at nucleotide position 118 (A118G). There have been many studies of the effects of this polymorphism on opioid dose requirements and response, although the studies in obstetric patients have had conflicting results. For example, laboring women who were AA homozygous had an increased intrathecal fentanyl requirement for analgesia.5 In contrast, AA homozygous women receiving intrathecal morphine for postcesarean analgesia reported less pain and required less patient-controlled morphine but had a higher incidence of nausea and vomiting.6 Wong7 summarized the conflicting evidence and noted that the true effect of this polymorphism is probably small. Many other genetic factors influence opioid disposition and response, and even more factors influence pain perception. Indeed, a meta-analysis found that A118G polymorphism only explained 7% of the variability in opioid requirements.8 Thus, at present there are no indications for pharmacogenetic testing in routine obstetric practice.9
Pharmacokinetic Changes
The major physiologic changes during pregnancy would be expected to alter drug disposition.10,11 However, the magnitude and time course of these changes vary throughout pregnancy and among individuals. The results of many older studies are unreliable because the studies were often of low quality. Thus, making generalizations about the effects of pregnancy on drug disposition can be difficult, and individualized dosing is important.
Maternal Pharmacokinetics
Absorption and Uptake.
Oral absorption and bioavailability are not usually affected by pregnancy, although nausea and vomiting may limit oral intake. Intestinal motility is decreased during pregnancy, but gastric emptying is only delayed during labor or after opioid administration. Cardiac output is increased by 30% to 50% during pregnancy, and the increased blood flow to skin and mucous membranes will enhance absorption from these sites. Reduced functional residual capacity and increased minute ventilation lead to increased pulmonary uptake of inhalational anesthetic agents.
Distribution.
The increased cardiac output during pregnancy increases distribution of drug to all tissues. Drugs acting peripherally (e.g., neuromuscular blockers) will be delivered to their site of action more quickly. However, the onset of intravenous and inhalational anesthetics is dependent on the time course of their cerebral drug concentrations. A delay in the increase in arterial and brain anesthetic concentrations will result from increased peripheral perfusion. Increased peripheral perfusion will, however, increase the return of drug during the elimination phase. Total body water increases on average by 8 L, and intravascular plasma volume is increased by 40%, whereas extravascular volume increases by a variable amount, depending on weight gain and edema. Thus, hydrophilic drugs, such as neuromuscular blockers, will have a small increase in the volume of distribution. Body fat is increased on average by 4 kg, but this is unimportant given the large volume of distribution of lipophilic drugs.
Changes in protein binding are more important clinically. Plasma albumin concentration is reduced to about 70% of normal, whereas α1-acid glycoprotein concentration is largely unchanged. Protein binding of drugs may be reduced by increased concentrations of free fatty acids and other endogenous displacing substances. This leads to increased concentrations of free drug, but with chronic drug administration this is offset by increased clearance of that free drug. The total (free + bound) concentration of drug will decrease, and it may be necessary to reset the therapeutic target range lower to compensate. Thus, it is important to know whether monitored concentrations are for free or total drug. Only a few drugs (e.g., theophylline, phenytoin) require monitoring and modification of dose because of changes in protein binding.
Metabolism.
Most drugs are metabolized in the liver, and the rate of metabolism may depend on hepatic blood flow or intrinsic enzyme activity. Although cardiac output is increased in pregnancy, it is not clear whether blood flow to the liver is significantly increased. Two studies using clearance of markers concluded that hepatic blood flow was unchanged,12,13 whereas another using Doppler ultrasonography reported unchanged hepatic arterial flow during pregnancy but increased portal venous flow after 28 weeks’ gestation.14 More importantly, some cytochrome P450 isoenzymes (CYP3A4, CYP2D6, and CYP2C9) and uridine diphosphate glucuronosyltransferase (UGT) isoenzymes (UGT1A4 and UGT2B7) have increased activity during pregnancy,10,15 which increases the metabolism of drugs such as phenytoin (CYP2C9), midazolam (CYP3A4), and morphine (UGT2B7). Other isoenzymes (CYP1A2 and CYP2C19) have decreased activity, which reduces the metabolism of drugs such as caffeine and theophylline (CYP1A2).
Elimination.
Renal blood flow is increased by 60% to 80% and glomerular filtration rate is increased by 50% in pregnancy; thus, the renal excretion of unchanged drugs such as cephalosporin antibiotics is increased. There is also increased activity of transporter proteins such as renal P-glycoprotein, which may contribute to the increased clearance of digoxin in pregnancy.16 Increased minute ventilation enhances elimination of inhalational anesthetic agents.
The physiologic changes of pregnancy will affect individual drugs depending on their physicochemical characteristics and metabolic pathways. Bioavailability is not usually changed significantly. Changes in volume of distribution as a result of changes in protein binding may affect drugs such as phenytoin, but monitoring and modification of therapy is usually straightforward. Drugs metabolized by the liver may require increases or decreases in dose, depending on the metabolic pathway involved. Drugs excreted unchanged by the kidneys often require an increased dose.
Placental Transfer and Metabolism
Our understanding of placental transfer and metabolism is rapidly improving (see Chapter 4). Early research was often limited to measuring drug concentrations in the umbilical vessels and maternal vein at delivery. Results were variable and difficult to interpret, especially for drugs such as anesthetic agents that are administered shortly before delivery. Umbilical blood samples are obtained at variable times after drug exposure, well before steady-state conditions are achieved. The theory of a placental barrier was proposed because maternal and fetal concentrations were often different. However, differences in concentrations of binding proteins are mainly responsible for the fetal-maternal distribution of drugs at steady state.17 The fetal concentration of albumin is slightly greater than in the mother, but α1-acid glycoprotein concentration is only a third of the maternal value at term. Umbilical-to-maternal blood ratios of total drug may be misleading because it is the free drug that equilibrates across the placenta. Maternal-to-fetal ratios of drugs do not provide information on the rate of drug transfer or the amount of drug that has already been transferred to the fetus.
Drug transfer across the placenta was previously thought to occur mainly by diffusion. This would favor the movement of lipophilic drugs, and placental perfusion would be an important factor affecting transfer. Fetal pH is lower than maternal pH, so that weak bases become more ionized in the fetus, thus limiting their transfer back across the placenta. Normally, the difference in pH is only 0.1 and this “ion trapping” is irrelevant, but fetal acidosis can significantly increase the fetal concentration of drugs such as local anesthetics.
It is now known that the placenta contains many drug transporters that can modify fetal drug exposure, and these are particularly relevant when trying to use transplacental pharmacotherapy to deliver drugs to the fetus.18,19 For example, in the treatment of sustained fetal tachyarrhythmia, placental P-glycoprotein, an adenosine triphosphate–dependent drug efflux pump, will reduce net transfer of substrates such as digoxin and verapamil from the mother. With maternal human immunodeficiency virus (HIV) infection, treatment of the fetus is also required, but drug transporters limit the transfer of some antiviral drugs, such as protease inhibitors.
The placenta also contains many enzymes, especially those such as UGT that catalyze phase II conjugation reactions.19,20 Clearance of substrates by UGT in full-term placentas may be sufficient to contribute to overall maternal metabolism.
Fetal and Neonatal Elimination.
The fetus and neonate metabolize drugs, but at a reduced rate compared with adults.21,22 The fetal circulation guides drug transferred across the placenta to undergo first-pass hepatic metabolism, but some drug will bypass the liver. Renal blood flow is minimal until near term, and any excreted products would just pass into the amniotic fluid to be swallowed. Elimination of drugs by the fetus is thus mainly reliant on placental transfer. It would seem prudent to minimize the amount of drug transferred to the neonate and choose drugs that are eliminated rapidly. Relatively large minute ventilation promotes neonatal elimination of inhalational anesthetic agents, and this may be further increased by assisted ventilation.
Pharmacodynamic Changes
Changes in the concentrations of various hormones may alter the response to other substances. In particular, progesterone and endorphins may enhance sedation and anti-nociception, respectively. A pharmacodynamic difference specifically refers to a change in response to a given effect-site concentration, but it is difficult during pregnancy to carry out the high-fidelity studies necessary for accurate pharmacokinetic-pharmacodynamic modeling. Thus, the demonstrations of pharmacodynamic changes in pregnancy have been limited to specific experimental designs where there are large differences in effect.
General Anesthesia
Early animal studies showed that maternal anesthetic requirements were reduced during pregnancy. Minimum alveolar concentration (MAC) values for inhalational agents were reduced by 25% to 40% in pregnant ewes23 and by 16% to 19% in pregnant rats.24 Ethical and practical difficulties with research in pregnant women delayed confirmation of this finding in humans. Isoflurane MAC (determined using transcutaneous electrical stimulation instead of the classic skin incision) was decreased by 28% in women undergoing termination of pregnancy at 8 to 12 weeks’ gestation.25 Similar reductions in MAC were found for enflurane (30%) and halothane (27%).26 MAC was reduced by 30% in the immediate postpartum period, with a return to nonpregnant values by 12 to 72 hours after delivery (Figure 14-1).27,28
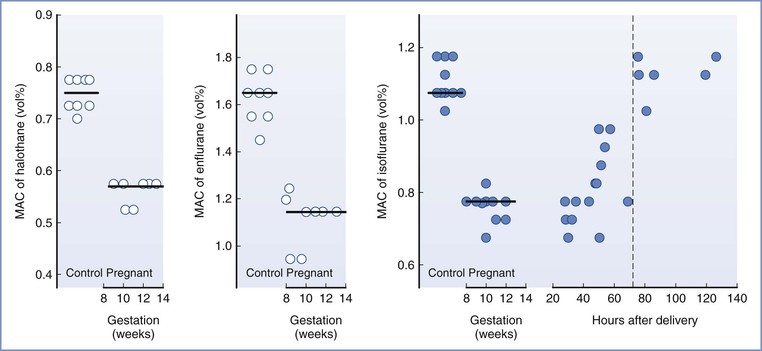
FIGURE 14-1 Changes in minimum alveolar concentration (MAC), determined by response to transcutaneous electrical stimulation, for halothane, enflurane, and isoflurane in early pregnancy and for isoflurane in the early postpartum period. (Reproduced with permission from Gin T. Obstetric pharmacology. In Evers AS, Maze M, Kharasch ED, editors. Anesthetic Pharmacology: Basic Principles and Clinical Practice, 2nd edition. Cambridge, Cambridge University Press, 2011:948-62. Original data from references 25, 26, and 27.)
Progesterone is probably the cause of the reduced anesthetic requirements during pregnancy; chronic progesterone administration reduced MAC in rabbits, dogs, and sheep.29–31 Although human studies have not found a good correlation between progesterone concentrations and the reduction in anesthetic requirement, a poor correlation may be expected if the effect of progesterone is not dose-dependent; it is possible that progesterone concentrations only need to exceed a low threshold to decrease anesthetic requirements. Lower concentrations of sevoflurane were required to maintain anesthesia in nonpregnant women during the luteal phase of the menstrual cycle, when progesterone concentrations are elevated, compared with during the follicular phase.32 The progesterone concentrations during pregnancy are much greater than those seen during the luteal phase of the menstrual cycle. The reduced MAC during pregnancy may also be a result of the increased endogenous endorphins that mediate the increase in nociceptive threshold during pregnancy; it is well known that opioids reduce MAC.
Pregnancy also alters other measures of anesthetic effect. In early pregnancy, the isoflurane concentration required for hypnosis was reduced by 31% and the bispectral index (BIS) was decreased at isoflurane concentrations over the range 0.1% to 2.0%.33 During the second trimester, the sevoflurane concentration required to achieve a targeted BIS of 50 was reduced by 31%.34 Both in early and term pregnancy, the median concentration of nitrous oxide required for loss of consciousness (MACawake) was reduced by 25% to 27%.35 One study did not show any difference in electroencephalographic (EEG) measures between women having cesarean delivery or gynecologic surgery, but there were many confounding factors such as the study being conducted partly during and partly after surgery, the concurrent use of significant doses of fentanyl, large variations in EEG measures, and small sample size.36
The data for intravenous anesthetic agents are more variable, partly because of methodologic challenges. It is difficult to produce a stable effect-site concentration of intravenous drugs to allow accurate measurement of drug effect. Increased cardiac output usually results in an increase in intravenous anesthetic dose requirements to produce central effects, and this change would counter any decrease in requirements with pregnancy. The bolus dose of thiopental for hypnosis (failure to open eyes to command) was 17% lower, and that for anesthesia (no purposeful movement to a transcutaneous electrical stimulus) was 18% lower in early pregnancy compared with nonpregnant women (Figure 14-2).37 A similar reduction was found in the early postpartum period, less than 60 hours after delivery.38
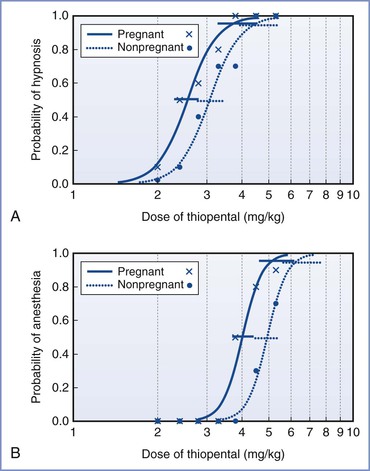
FIGURE 14-2 Calculated dose-response curves (log[dose] scale) for thiopental for hypnosis (A) and anesthesia (B) in pregnant and nonpregnant women. The 95% confidence intervals for the values of ED50 and ED95 are also displayed, slightly offset for clarity. Raw data are shown for pregnant women (x) and nonpregnant women (•). (Modified with permission from Gin T, Mainland P, Chan MTV, Short TG. Decreased thiopental requirements in early pregnancy. Anesthesiology 1997; 86:73-8.)
Studies using target-controlled infusions may not be reliable because the pharmacokinetic models may not be accurate in pregnancy, and they are known to predict concentrations poorly at induction of anesthesia. These methodologic problems may be the reason one study found no differences in the concentration of propofol required for loss of consciousness in early pregnancy.39 Another study used a slow infusion of propofol for induction of anesthesia and found that the dose and calculated effect-site concentrations at loss of consciousness were 8% lower than in nonpregnant women.40 The reduction in anesthetic requirement for intravenous agents appears to be less (8% to 18%) than that for inhalational agents (approximately 30%). It is not known whether this reflects real differences between the drugs or the methodologic problems just outlined.
Local Anesthesia
The spread of neuraxial block is increased in pregnant women (see Chapter 2). This has been shown as early as the first trimester for epidural anesthesia41 and the second trimester for spinal anesthesia.42 One small study suggested that although the spread of epidural block was increased, the latency and density of sensory and motor block were not.43 However, two more recent studies showed that the median effective dose of intrathecal bupivacaine for motor block was decreased by 13% to 35% in pregnant women at term.44,45 Magnetic resonance imaging has confirmed that pregnant women have increased epidural blood volume, decreasing the capacity of the epidural space and decreasing the volume of lumbar cerebrospinal fluid.46,47 These mechanical factors would explain the increased spread of local anesthetic. However, several studies have also shown that there is increased sensitivity to local anesthetics during pregnancy. The onset of conduction block in the vagus nerve with bupivacaine was faster in pregnant versus nonpregnant rabbits.48,49 Sciatic nerve block was of longer duration and the lidocaine content in the nerves was lower at the time of return of deep pain in pregnant versus nonpregnant rats.50 Sensory nerve action potentials were inhibited to a greater extent during median nerve block at the wrist with lidocaine in pregnant versus nonpregnant women.51 The increased sensitivity may be caused by progesterone because exogenously administered progesterone increased the susceptibility of rabbit vagus nerves to bupivacaine.52 One study found no changes in conduction block in pregnant rats and suggested that enhanced block may be due to pregnancy-induced changes that facilitate diffusion of local anesthetic or an interaction with endogenous analgesic systems.53
Analgesia
Pregnancy is associated with increases in nociceptive response thresholds that are mediated by endogenous opioid systems.54,55 The changes in threshold can be reproduced using exogenous progesterone and estrogen and appear to involve spinal cord kappa (κ) and delta (δ) opioid receptors and descending spinal α2-noradrenergic pathways.56 Early human studies produced mixed results, probably because of methodologic problems. Recent controlled studies showed that heat pain threshold was increased in term pregnant women, and this persisted during the first 24 to 48 hours after delivery.57,58 Given the many different factors that influence pain behavior, especially those unique to pregnancy and delivery, it is difficult to determine how this change in pain threshold influences perioperative analgesic requirements.
Drug Use during Pregnancy
General Teratology
Teratology is the study of abnormal development or birth defects. Teratogens are substances that act to irreversibly alter growth, structure, or function of the developing embryo.59 Ideally, preclinical studies would identify teratogens, but drug teratogenicity unfortunately can be markedly species-specific. For example, thalidomide produces phocomelia in primates but not in rodents.
In the United States, major malformations affect 2% to 3% of neonates.60 A major malformation is defined as one that is incompatible with survival (e.g., anencephaly), one that requires major surgery for correction (e.g., cleft palate, congenital heart disease), or one that causes mental retardation. If all minor malformations (e.g., ear tags, extra digits) are included, the incidence of congenital anomalies may be as high as 7% to 10%. Exogenous causes of birth defects (e.g., radiation, infections, maternal metabolic disorders, drugs, environmental chemicals) account for almost 10% of all major birth defects and therefore affect only 0.2% to 0.3% of all births. Drug exposure explains only 2% to 3% of birth defects, and the majority of birth defects are of unknown etiology.
To avoid unnecessary and potentially teratogenic exposures, nonpharmacologic techniques should be used when possible and drugs should be used only when necessary. The risk-to-benefit ratio should justify the use of a drug given to a pregnant woman, and the minimum effective dose should be employed. Long-term effects of fetal drug exposure may not become apparent for many years. Therefore, physicians and patients should exercise caution in the use of any drug during pregnancy. On the other hand, the physician should ask the following question: what would be the appropriate treatment in the nonpregnant patient with the same condition? In most cases, the answer is the same as that for women who are pregnant.61
Sensitive serum pregnancy tests can diagnose pregnancy as early as 1 week after conception. Before drug therapy is started, a sensitive test should be used if there is any question about drug safety during a potential pregnancy.
It is also important to remember that the male partner may be taking teratogenic drugs and the drug may be present in semen at low concentrations. Although the magnitude of fetal risk is unclear, men are advised to avoid drugs such as thalidomide, ribavirin, and isotretinoin if their partners could become pregnant.
The maternal and fetal genotype and phenotype can affect individual susceptibility to an agent. For example, fetuses with low levels of the enzyme epoxide hydrolase are more likely to manifest fetal hydantoin syndrome than those with normal levels of this enzyme.62
Drug teratogenicity is affected by the timing of exposure. Teratogen exposure in the first 2 weeks after conception is generally thought to be an all-or-nothing phenomenon (i.e., having either no effect or resulting in spontaneous fetal loss). Among women with a 28-day menstrual cycle, the classic period of susceptibility to teratogenic agents is during the period of organogenesis, which occurs primarily at 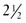 to 8 weeks after conception (31 to 71 days, or 4 to 10 weeks, after the first day of the last menstrual period) (Figure 14-3). During organogenesis, each organ system has different critical periods of sensitivity and there may be striking differences in effect. When administered between 35 and 37 days’ gestation, thalidomide produces ear malformations; when administered between 41 and 44 days’ gestation, it produces amelia or phocomelia. After this period, embryonic development is characterized primarily by increasing organ size; thus, the principal effect of exposure consists of growth restriction and/or effects on the nervous system and gonadal tissue. For example, diethylstilbestrol exposure during the second trimester results in uterine anomalies that do not become apparent until after puberty. Fetal alcohol syndrome may occur with chronic exposure to alcohol during pregnancy.
to 8 weeks after conception (31 to 71 days, or 4 to 10 weeks, after the first day of the last menstrual period) (Figure 14-3). During organogenesis, each organ system has different critical periods of sensitivity and there may be striking differences in effect. When administered between 35 and 37 days’ gestation, thalidomide produces ear malformations; when administered between 41 and 44 days’ gestation, it produces amelia or phocomelia. After this period, embryonic development is characterized primarily by increasing organ size; thus, the principal effect of exposure consists of growth restriction and/or effects on the nervous system and gonadal tissue. For example, diethylstilbestrol exposure during the second trimester results in uterine anomalies that do not become apparent until after puberty. Fetal alcohol syndrome may occur with chronic exposure to alcohol during pregnancy.
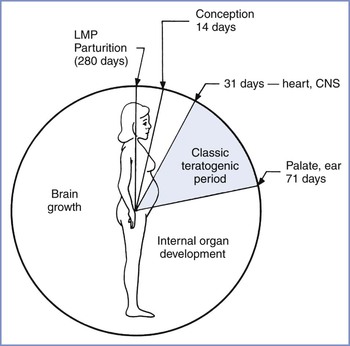
FIGURE 14-3 Gestational clock showing the classic teratogenic period. CNS, central nervous system; LMP, day of last menstrual period. (From Niebyl JR. Drug Use in Pregnancy. 2nd edition. Philadelphia, Lea & Febiger, 1988:2.)
The drug dosing regimen can influence teratogenicity. In most cases, administration of a low dose has no effect whereas malformations may occur at intermediate doses and death may occur at higher doses. Fetal death may allow organ-specific teratogenic activity to go unnoticed. Small doses administered over several days may have an effect different from that observed with the same total dose administered at one time. Sequential drug administration may induce the production of an enzyme that metabolizes the drug and thus results in less exposure. Constant exposure may destroy cells that would have catabolized the drug if it had been administered in periodic doses. Combinations of agents may produce different degrees of malformation and growth restriction from those that occur with drugs administered individually. For example, fetuses whose mothers receive combination anticonvulsant therapy are at the highest risk for malformations, including neural tube defects and facial dysmorphic features.
U.S. Food and Drug Administration Categories
In 1979, the U.S. Food and Drug Administration (FDA) introduced a drug classification system to discourage nonessential use of medications during pregnancy (Box 14-1).
Unfortunately, maternal anxiety related to medication use can lead to unnecessary pregnancy terminations. Several characteristics of the FDA drug classification system contribute to public perception—and misperception—of the dangers of using medication during pregnancy. Although only 20 to 30 commonly used drugs are known teratogens, 7% of all the medications that are listed in the Physicians’ Desk Reference are classified as Category X.63,64 All new medications are classified as Category C, leading to an exaggerated impression of the danger of many medications. In addition, the manufacturer’s prescribing information for many drugs may state that the drug is not approved for use in pregnancy despite a long history of uncomplicated unlicensed or “off-label” use.65
The FDA categories imply a progressive fetal risk from Category A to X; however, the drugs in different categories may pose similar risks but may be listed in different categories on the basis of risk-to-benefit considerations. In addition, the categories create the impression that drugs within a category present similar risks whereas the category definition permits inclusion (in the same category) of drugs that vary in type, degree, and extent of risk.
Use of potentially teratogenic drugs in pregnancy is surprisingly commonplace. Over 63% of pregnant patients in Canada filled a prescription for at least one drug, with almost 8% filling a prescription for a Category D or X medication.66
When counseling patients or responding to queries from physicians, we prefer to avoid referring to the Physicians’ Desk Reference. Rather, we use specific descriptions in teratogen databases to provide the best information available. Many resources are freely available online, in addition to the commercially available databases (Table 14-1).
TABLE 14-1
Internet Resources for Additional Drug and Teratogen Information
| American Academy of Pediatrics: The Transfer of Drugs and Other Chemicals into Human Milk | http://pediatrics.aappublications.org/content/108/3/776.full.html |
| The American Botanical Council | http://www.herbalgram.org |
| The American College of Obstetricians and Gynecologists | http://www.acog.org/About_ACOG/ACOG_Departments/Resource_Center/WEBTREATS_Teratology_Toxicology |
| Motherisk | http://www.motherisk.org |
| Organization of Teratology Information Specialists: Fact sheets on exposure during pregnancy to a variety of diseases, medications, and herbal remedies | http://otispregnancy.org/otis_fact_sheets.asp |
| The National Library of Medicine PubMed | http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed |
| The National Institutes of Health National Center for Complementary and Alternative Medicine | http://nccam.nih.gov |
| The National Institutes of Health Office of Dietary Supplements | http://dietary-supplements.info.nih.gov |
| Perinatology.com: Drugs in Pregnancy and Breastfeeding | http://www.perinatology.com/exposures/druglist.htm |
| The Reproductive Toxicology Center* | http://www.reprotox.org |
| RxList: The Internet Drug Index | http://www.rxlist.com |
| SafeFetus.com | http://www.safefetus.com |
| University of Washington Clinical Teratology Web* | http://depts.washington.edu/~terisweb |
| U.S. Food and Drug Administration Office of Women’s Health | http://www.fda.gov/AboutFDA/CentersOffices/OC/OfficeofWomensHealth/ |
| Drugs and Lactation Database (LactMed) | http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT |
* Databases, including Reprotox, Reprotext, Teris, and Shepard’s Catalog of Teratogenic Agents, can be purchased from these websites.
The Teratology Society has suggested abandonment of the FDA classification scheme.63 In 1997, the FDA held a public meeting to discuss labeling of drugs. There was consensus that the current classification scheme is probably oversimplified and confusing, does not address the range of clinical situations or the range of possible effects, and should be replaced with narrative labeling. Subsequently, a concept paper was presented that outlined a new model for labeling and included sections such as “clinical management statement,” “summary risk assessment,” and “discussion of data” for both pregnant and breast-feeding women.67 This proposal has not yet been implemented. The FDA Office of Women’s Health has created a pregnancy registry website, which lists a variety of registries that women who have used specific medications during pregnancy can consult (see Table 14-1). In 2008, the FDA stated they will eliminate the A, B, C, D, X classification system.68 As of 2013, the new system has yet to be implemented despite completion of the 90-day comment period some time earlier.69
Analgesics
There is no known teratogenic risk associated with the use of acetaminophen (paracetamol),70 which is the preferred mild analgesic or antipyretic during pregnancy.
Nonsteroidal anti-inflammatory drugs (NSAIDs) have not been associated with an increased risk for birth defects overall, but the occasional review has suggested an association with specific defects.71 For example, aspirin is not associated with an overall increase in rates of congenital malformations, but one review suggested a higher risk for gastroschisis.72 Aspirin is not usually the first choice of NSAID, but low-dose aspirin may still have a role in some situations, such as preventing fetal loss associated with antiphospholipid antibody syndrome.73 Studies of the use of NSAIDs and aspirin in the first trimester have reported increased risk for pregnancy loss (adjusted odds ratio [OR], 1.8 to 8.1).71 In the second trimester, their use has been associated with fetal cryptorchidism. In the third trimester, NSAIDs and aspirin are usually avoided because of significant fetal risks, such as renal injury, oligohydramnios, and intrauterine constriction of the ductus arteriosus, an effect that increases with advancing gestational age.74 Renal injury, necrotizing enterocolitis, and intracranial hemorrhage are other potential complications.
Opioids such as propoxyphene and codeine have no known teratogenic risk,75 but they have well-known potential for addiction. Excessive antepartum use can also lead to neonatal opioid-withdrawal symptoms.76 Surprisingly, a recent case-control study found that maternal opioid treatment (mostly codeine and hydrocodone) between 1 month before pregnancy and the first trimester was associated with an increased OR of 1.8 to 2.7 for various cardiac birth defects, spina bifida, and gastroschisis.77
Tramadol has analgesic effects from weak opioid activity and inhibition of serotonin and norepinephrine uptake. Despite availability in some countries for more than 30 years, few data are available regarding potential adverse effects in pregnancy. Tramadol exposure in early pregnancy was associated with a higher number of spontaneous abortions, and it should be avoided in the first trimester.78 Chronic tramadol use in later pregnancy may result in neonatal withdrawal syndrome.
Sedatives
Human epidemiologic studies of the possible teratogenic effects of various tranquilizers are inconsistent. One report of nearly 400 patients found a 12% incidence of birth defects in the offspring of meprobamate users.79 Another study of similar size did not identify any higher risk for malformations.80 These latter two articles found similar results for chlordiazepoxide.79,80 A recent study evaluating meprobamate used in high doses to attempt suicide did not show any teratogenic or fetotoxic effects.81 A sample of 35 pregnant women who self-poisoned with chlordiazepoxide showed no association with congenital abnormalities but did show dose-related fetal growth restriction.82
Some studies have suggested that first-trimester exposure to diazepam increases the risk for cleft lip with or without cleft palate, neural tube defects, intestinal atresia, and limb defects.83 Other reports have not suggested an increase in rate of congenital abnormalities after fetal exposure to benzodiazepines. In a case-control study of 611 infants with cleft lip or cleft palate and 2498 controls with other birth defects, after adjustment of the data for potential confounders, no association between diazepam and cleft palate was found (OR, 0.8 for cleft lip with or without cleft palate, with 95% confidence interval [CI] of 0.4 to 1.7, and OR, 0.8 for cleft palate alone, with 95% CI of 0.2 to 2.5).84 Reanalysis of the Hungarian Case-Control Surveillance of Congenital Abnormality data also showed a weak relationship between exposure to diazepam in early pregnancy and neural tube defects, limb deficiency defects, and possibly intestinal atresia or stenosis.83 One study found no difference in the incidence of congenital anomalies between 460 women exposed to benzodiazepines during pregnancy and 424 control women without such exposure (i.e., 3.1% versus 2.6%, respectively).84 Perinatal use of diazepam has been associated with hypotonia, hypothermia, and respiratory depression.85 Overall, it appears that the teratogenic risk of benzodiazepines is small at most,86,87 but there may be a small risk for preterm birth and low birth weight.88 Benzodiazepines should only be used in the first trimester if the perceived benefit offsets the possible teratogenic risks and later neonatal effects of continued use. Some benzodiazepines such as temazepam are classified as FDA Category X.
Anticonvulsants
Epilepsy is the most common serious neurologic problem during pregnancy.89 It has been estimated that 3 to 5 births per thousand will be to women with epilepsy.90 All anticonvulsants cross the placenta. The fetal congenital anomaly rate in pregnant women with epilepsy who ingest anticonvulsant drugs is 4% to 8%, compared with a background incidence in the general population of 2% to 3%.91,92 A twofold higher risk for minor malformations also exists in this population.92 Cleft lip, with or without cleft palate, and congenital heart disease are especially common. Administration of valproic acid or carbamazepine entails a 1% risk for neural tube defects and other malformations; thus, alpha-fetoprotein screening and targeted ultrasonography are appropriate for patients taking these agents. In addition, the offspring of epileptic women have a 2% to 3% incidence of epilepsy, which is five times that of the general population.
Holmes et al.93 attempted to refute the unproven theory that women with epilepsy have a genetic propensity to have children with a higher risk for birth defects that is separate from the greater risk associated with the use of anticonvulsants. These investigators studied children of women who had a history of seizures but took no medications during the pregnancy. There was no difference in physical features or cognitive function between these children and a group of matched controls. In a second study, Holmes et al.94 screened 128,049 pregnant women at delivery to identify the following three groups of infants: (1) those exposed to anticonvulsant drugs, (2) those unexposed to anticonvulsant drugs but with a maternal history of seizures, and (3) those unexposed to anticonvulsant drugs with no maternal history of seizures (control group). The frequency of anticonvulsant embryopathy was higher in the 223 infants exposed to one anticonvulsant drug than in the 508 control infants (20.6% versus 8.5%, respectively; OR, 2.8; 95% CI, 1.1 to 9.7). The frequency was also higher in 93 infants exposed to two or more anticonvulsant drugs than in the controls (28.0% versus 8.5%; OR, 4.2; 95% CI, 1.1 to 5.1). The 98 infants whose mothers had a history of epilepsy but took no anticonvulsant drugs during pregnancy did not have a higher frequency of abnormalities than the control infants. The investigators concluded that “a distinctive pattern of physical abnormalities in infants of mothers with epilepsy is associated with the use of anticonvulsant drugs during pregnancy, rather than with epilepsy itself.” A more recent meta-analysis also did not support the view that epilepsy per se represents a teratogenic risk.95
Possible causes of these congenital malformations include genetic differences in drug metabolism, the specific drugs themselves, and deficiency states (e.g., decreased folate levels) induced by the drugs. No congenital malformations appear to be unique to any one anticonvulsant. The characteristics of these syndromes are so similar that the broad term fetal anticonvulsant syndrome, consisting primarily of orofacial, cardiovascular, and digital malformations, has been applied to almost every anticonvulsant drug.96
Among women taking phenytoin, there is a 2% to 5% risk for major congenital anomalies, primarily midline heart defects, orofacial clefts, and urogenital defects.91 Fetal hydantoin syndrome is a constellation of minor anomalies, such as craniofacial abnormalities (short nose, flat nasal bridge, wide lips, hypertelorism, ptosis, epicanthal folds, low-set ears, and low hairline) and limb anomalies (distal digital hypoplasia, absent nails, and altered palmar crease). In addition, neonatal growth and performance delays have been documented. The risk for fetal hydantoin syndrome for the child of a woman taking phenytoin is approximately 10%.94 Phenytoin may act as a competitive inhibitor of the placental transport of vitamin K. This results in a decrease in fetal coagulation factors II, VII, IX, and X. In addition, phenytoin may induce fetal hepatic metabolism of the coagulation factors. The resulting reduction in fetal coagulation factors is associated with a higher risk for hemorrhagic disease of the newborn.97 To help prevent this coagulopathy, some physicians advocate oral vitamin K supplementation (10 mg daily) for pregnant epileptic patients during the last month of pregnancy in addition to the parenteral administration of vitamin K to the neonate at birth.98 Several anticonvulsant medications have metabolites that typically are eliminated by the enzyme epoxide hydrolase. In one study, 19 women taking phenytoin underwent amniocentesis. All 4 of the women with low enzyme activity in amniocytes had affected fetuses. The 15 fetuses with normal amniocyte epoxide hydrolase activity did not have the characteristics of fetal hydantoin syndrome.62
Carbamazepine is used to treat all types of seizure disorders, with the exception of petit mal epilepsy. It is most commonly used in the treatment of psychomotor (temporal lobe) epilepsy and grand mal epilepsy. In a prospective study involving 72 women with epilepsy who were taking carbamazepine, the incidence of congenital anomalies was higher in the 35 fetuses exposed only to this drug. There was an 11% incidence of craniofacial defects, a 26% incidence of fingernail hypoplasia, and a 20% incidence of developmental delay.99 This constellation of fetal effects, named fetal carbamazepine syndrome, closely resembles the malformations seen in cases of fetal hydantoin syndrome. In addition, maternal carbamazepine exposure has been specifically associated with spina bifida. An analysis of all available data involving cohorts of pregnant women ingesting carbamazepine supports the conclusion that fetal exposure to this drug carries a 0.5% to 1% risk for spina bifida.100 Although it is generally agreed that the use of carbamazepine in pregnancy is associated with a risk for neural tube defects and other anomalies, the exact magnitude of the risk from use of carbamazepine alone is unclear.101–104
Phenobarbital is used in the treatment of partial and generalized tonic-clonic seizures and status epilepticus.105 Fetal exposure to phenobarbital has been associated with major malformations, such as congenital heart defects and orofacial clefting. Fetal phenobarbital syndrome is characterized by minor dysmorphic features similar to those seen with fetal hydantoin syndrome.91
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


