However, in all species studied, including humans, the log of basal metabolic rate (BMR) plotted against the log of body weight produces a straight line with a slope of ¾. This is different to the BSA, which is described using body weight with an exponent of 2/3 [77]. Fractal geometry is used to mathematically explain this phenomenon known as allometry [78]. A great many physiological, structural, and time-related variables scale predictably within and between species with weight (W) exponents (PWR) of 3/4, 1, and 1/4, respectively [76].
These exponents have applicability to pharmacokinetic parameters such as clearance (CL), volume (V), and half-time [76]. The factor for size (F size) for total drug clearance may be expected to scale weight with an exponent of ¾:
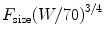 The use of these allometric models allows prediction of dose in children from that of adult dose; such prediction is not possible using the mg/kg scaling. Pediatric doses in children out of the neonatal age group are greater than adults.
The use of these allometric models allows prediction of dose in children from that of adult dose; such prediction is not possible using the mg/kg scaling. Pediatric doses in children out of the neonatal age group are greater than adults.
Remifentanil clearance in children 1 month–9 years is similar to adult rates when scaled using an allometric exponent of ¾ [79]. Remifentanil is hydrolyzed by nonspecific tissue and plasma esterases that do not appear to be influenced by age after scaling for size. Plasma esterases responsible for clearance are mature at birth [80].
The effect of size on the dose of remifentanil tolerated during spontaneous ventilation under anesthesia has been investigated in children undergoing strabismus surgery (n = 45, age 6 months–9 years). The propofol infusion was titrated using state entropy as a pharmacodynamic endpoint and remifentanil infused, using a modified up-and-down method, with respiratory rate depression as a pharmacodynamic endpoint. A respiratory rate of just greater than 10, stable for 10 min, determined the final remifentanil infusion rate [81]. This influence of age on the remifentanil infusion requirement is shown in Fig. 11.1 [81–83]. Superimposed on this figure are clearance estimates for age, determined by size using an allometric model with a standardized clearance of 2,790 mL/min for a 70 kg person. Clearance mirrors infusion rate in children over the age of 1 year. There is a divergence between clearance estimate and infusion rate in those children in infancy. The higher infusion rates recorded in those infants can be attributed to greater suppression of respiratory drive in this age group than the older children during the study; a respiratory rate of ten breaths per minute in an infant is disproportionately slow compared to the same rate in a 7-year-old child, suggesting excessive dose.
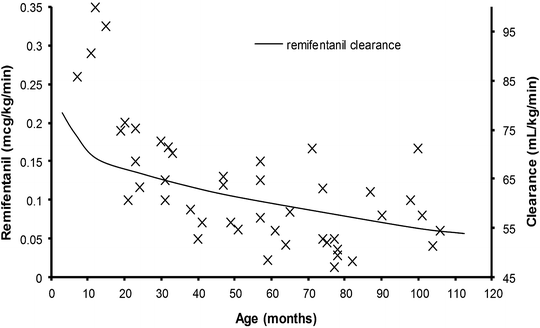
Fig. 11.1
The effect of age on the dose of remifentanil tolerated during spontaneous ventilation under anesthesia in children undergoing strabismus surgery [81]. Superimposed on this plot is estimated remifentanil clearance determined using an allometric model [82]. There is a mismatch between clearance and infusion rate for those individuals still in infancy (Reprinted with permission from Anderson BJ. Pediatric models for adult target-controlled infusion pumps. Paediatr Anaesth 2010 Mar;20(3):223-32)
For most drugs, however, allometry alone is insufficient to predict clearance in neonates and infants from adult estimates. Most clearance systems are not mature at birth. The addition of a model describing clearance maturation with age is required. The sigmoid hyperbolic function (also known as the Hill equation) [84] (also used to describe the oxygen saturation curve) has also been found useful for describing this maturation process (MF).
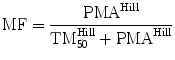 The TM50 describes the maturation half-time, while the Hill coefficient relates to the slope of this maturation profile. Maturation of a considerable number of drugs has now been described using this equation [85]. The maturation profile for dexmedetomidine expressed using allometric scaling, and this maturation model is shown in Fig. 11.2 [86].
The TM50 describes the maturation half-time, while the Hill coefficient relates to the slope of this maturation profile. Maturation of a considerable number of drugs has now been described using this equation [85]. The maturation profile for dexmedetomidine expressed using allometric scaling, and this maturation model is shown in Fig. 11.2 [86].
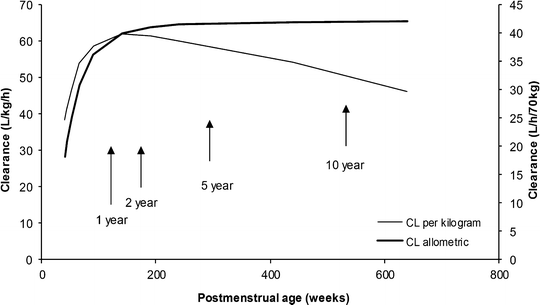
Fig. 11.2
Dexmedetomidine clearance changes with age, expressed both as per kilogram and using allometric scaling with a maturation model. The per kilogram model (L/h/kg) demonstrates an increased clearance in infants that explains the observed increased infusion (mg/min/kg) required for sedation in this age group. The use of the allometric model allows better understanding of the clearance maturation process (Data from Potts et al. [86])
Organ function (OF) remains the other major covariate influence on clearance. While renal pathology may be reflected by assessment such as creatinine clearance, distinguishing this from normal physiology in infants may be difficult unless ordinary renal maturation is understood [75]. Although specific organ dysfunction of the kidney or liver is well recognized as having effect on clearance, other processes (sepsis, malnutrition, disease severity scores) can also be used as markers of reduced clearance. Midazolam clearance was noted to be reduced in critically ill children. This was assumed to be due to reduced CYP3A activity, although reduced hepatic blood flow is more likely [87]. Pharmacokinetic parameters (P) can be described in an individual as the product of size (F size), maturation (MF), and organ function (OF) influences where P std is the value in a standard size adult without pathological changes in organ function:
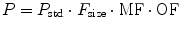 This methodology is increasingly used to describe clearance changes with age [85]. An understanding of these principles can be used to predict dose in children using target concentration methodology [88].
This methodology is increasingly used to describe clearance changes with age [85]. An understanding of these principles can be used to predict dose in children using target concentration methodology [88].
When maturation changes have not been described using real data, then an alternative method known as the physiological-based pharmacokinetic (PBPK) model can be used to predict changes with age. Organ maturation, body composition, and ontogeny of drug elimination pathways have marked effects on pharmacokinetic parameters in the first few years of life. PBPK models require detailed physiological data. Data on ontogeny of individual clearance pathways, derived from measurements of enzyme expression and activity in postmortem livers, and from in vivo data from drugs that are cleared by similar pathways are useful. Continued input of information concerning genetic, physiological, organ and tissue size and composition, protein binding, demographic and clinical data into the library, and algorithms for PBPK modeling programs has progressively improved their prediction ability. These models have been used to assist with first-time dosing in children [89–91]. The introduction of population variability in enzyme abundance and activity contributes to between-individual variability estimates [92]. This approach has been recently used to investigate fentanyl maturation changes with age in neonates [93].
Hepatic Elimination
Phase 1
The mixed function P450 (CYP, so named because these enzymes absorb light at wavelengths near 450 nm) oxidases are reduced [94, 95]. Enzyme maturation occurs at different rates. Individual hepatic drug-metabolizing enzymes have been categorized into one of three classes based on developmental trajectories [96]. Class 1 enzymes are expressed at their highest levels in the fetus during the first trimester (e.g., CYP3A7 that may have a role in retinoic metabolism); enzymes belonging to the second class are expressed at relatively constant levels throughout gestation and into adulthood (e.g., sulfotransferase, SULT1A1, responsible for most of paracetamol clearance in neonates). The vast majority of enzymes (Class 3, e.g., CYP3A4, UGT2B7; see below) are expressed at low levels at birth and mature with time.
The activity of the CYP2E1 enzyme surges after birth [97], CYP2D6 (e.g., codeine, tramadol) becomes detectable soon thereafter, and the CYP3A4 (e.g., midazolam) and CYP2C (e.g., diclofenac) family appear during the first week, whereas CYP1A2 (e.g., caffeine) is the last to appear [98]. Neonates are dependent on the immature CYP3A4 for levobupivacaine clearance and CYP1A2 for ropivacaine clearance, dictating reduced epidural infusion rates in this age group [99–101]. Some of these maturation rates have been described, e.g., midazolam [102] and levobupivacaine [103].
If a drug has a high extraction ratio, then intrinsic clearance may be very much greater than liver blood flow, and in these situations, hepatic clearance is primarily determined by liver blood flow characteristics. Fentanyl clearance (CYP3A4) is 70–80 % of adult values in term neonates and, standardized to a 70-kg person, reaches adult values within the first few weeks of life [57]. Omphalocele repair may be associated with raised intra-abdominal pressure (an organ function effect) resulting in reduced fentanyl clearance attributable to decreased hepatic blood flow.
Phase 2
Some phase II pathways are mature in term neonates at birth (sulfate conjugation), while others are not (acetylation, glycination, glucuronidation) [104]. Allometric body-size scaling complimented by maturation models [76, 105] has been used to unravel the developmental PK of drugs cleared by glucuronosyltransferase. Paracetamol and morphine are cleared by individual isoforms of glucuronosyltransferase (UGT1A6 and UGT2B7), as is bilirubin (UGT1A1). Clearance of both drugs [106–108] is immature in the premature 24-week PMA neonate and mature to reach adult rates by the first year of life. Dexmedetomidine is also cleared predominantly by the UGT system and has a similar maturation profile [109]. Glucuronidation is also the major metabolic pathway of propofol metabolism, although multiple cytochrome P450 isoenzymes, including CYP2B6, CYP2C9, or CYP2A6, contribute to its metabolism and cause a faster maturation profile (Fig. 11.3) [105, 109–113] than expected from glucuronide conjugation alone [73].
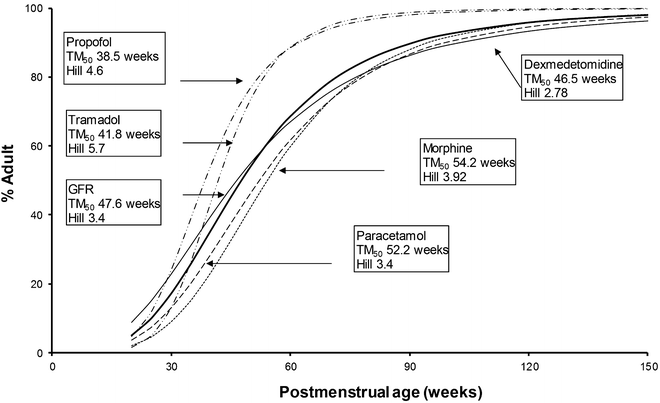
Fig. 11.3
Clearance maturation, expressed as a percentage of mature clearance, of drugs where glucuronide conjugation (paracetamol, morphine, dexmedetomidine) plays a major role. These profiles are closely aligned with glomerular filtration rate (GFR). In contrast, cytochrome P450 isoenzymes also contribute to propofol metabolism and cause a faster maturation profile than expected from glucuronide conjugation alone. Tramadol clearance maturation (phase I, CYP2D6, CYP3A) is also rapid. Maturation parameter estimates were taken from references [105, 109–113]
This maturation process can be difficult to discern because other factors such as illness impact on observed clearance. Morphine clearance is greater in infants undergoing noncardiac surgery than in those after cardiac surgery [114], or in those receiving extracorporeal membrane oxygenation [115] or positive pressure ventilation [110]. Similarly clearance of propofol was reduced after cardiac surgery in children admitted to a pediatric intensive care [116]. Dexmedetomidine clearance is reduced with low cardiac output, consistent with reduced hepatic blood flow [117]. A circadian night rhythm effect was noted in an investigation of infant propofol sedation after major craniofacial surgery [118].
Renal Elimination
Drugs and their metabolites are excreted by the kidneys by two processes—glomerular filtration and tubular secretion—that mature at different rates [119]. Glomerular filtration rate (GFR) is only 10 % that of mature value at 25 weeks, 35 % at term, and 90 % of the adult GFR at 1 year of age [111]. Tubular secretion maturation lags behind that of GFR [119]. Aminoglycosides are almost exclusively cleared by renal elimination, and maintenance dose is predicted by PMA because it predicts the time course of development of renal function [120]. The clearance of the old NMBD, d-tubocurarine, can be directly correlated with GFR [121].
Immaturity of clearance pathways can be used to our advantage when managing apnea after anesthesia in the premature nursery graduate. N7-methylation of theophylline in the newborn to produce caffeine is well developed whereas oxidative demethylation (CYP1A2) responsible for caffeine metabolism is deficient and develops over the ensuing months. Theophylline is effective for the management of postoperative apnea in the premature neonate, partly because it is a prodrug of caffeine, which is effective controlling apnea. Caffeine can only be slowly cleared by the immature kidney and is mature by 60 weeks PMA [122].
Pulmonary Elimination
The factors determining anesthetic absorption (alveolar ventilation, FRC, cardiac output, tissue/blood solubility) also contribute to elimination. We might anticipate more rapid washout in neonates than adults for any given duration of anesthesia because there is less distribution to fat and muscle content. The greater decrease in cardiac output induced by halothane in neonates might be expected to speed elimination, but brain perfusion will also be reduced and this slows recovery. Halothane, in particular, and to a far lesser extent isoflurane and sevoflurane undergo hepatic metabolism, but contribution is small compared to pulmonary elimination [123].
Metabolites
Many drugs have active metabolites that contribute to effect. Examples include norketamine from ketamine [124], 4′-hydroxydiclofenac from diclofenac [125], O-demethyl tramadol from tramadol [112], hydroxymidazolam from midazolam [126], and morphine 6-glucuronide (M6G) from morphine [127].
Contributions to both the desired effect (analgesia) and the undesired effects (nausea, respiratory depression) of M6G remain uncertain [128]. The EC50 for both morphine and M6G appear similar [129, 130], but M6G takes greater time (4–8 h versus 16 min) to equilibrate with the effect site [129, 131]. The relative ratios of morphine to M6G vary in neonates and early infancy, depending on relative maturation of UGT2B7 (formation of M6G) and GFR (elimination of M6G). Term neonates less than 7 days old have a lower ratio of plasma morphine/M6G than those over 1 year despite similar doses [74, 132]. The impact of this is unclear.
Pharmacogenomics
Pharmacogenomics (PG) is the investigation of variations of DNA and RNA characteristics as related to drug response that incorporates both PK and PD. There is large between-individual PK variability that is contributed to by polymorphisms of the genes encoding for metabolic enzymes [133]. Genetic variability influencing plasma cholinesterase activity and its influence on succinylcholine is a well-known example. Another example is the CYP2D6 single nuclear polymorphism (SNP) that is inherited as an autosomal recessive trait. Homozygous individuals are deficient in the metabolism of a variety of important groups of drugs: β(beta)-adrenoceptor blocking agents, antidepressants, neuroleptic agents, and opioids. Poor metabolizers have reduced morphine production from codeine [134, 135]. Tramadol is also metabolized by O-demethylation in the liver (CYP2D6) to O-desmethyl tramadol (M1), and the M1 metabolite has a mu-opioid affinity approximately 200 times greater than tramadol.
An SNP is only important if it contributes greater than 50 % metabolism, has an active metabolite, a steep dose–response relationship, and a narrow therapeutic index. These polymorphisms may have little impact during the neonatal period when metabolism is developmentally limited [5, 112, 136–138]. SNPs will certainly have impact in infants and children. Impact will be dependent on the rate of maturation of the specific enzyme system. Certainly ultra-extensive metabolizers of codeine (CYP2D6) may suffer respiratory depression resulting from the rapid formation of morphine [139]. Reports of death due to respiratory compromise in children given codeine for pain relief after tonsillectomy may limit the future use of this drug [140].
Pharmacogenomic differences also have impact on PD. Candidate genes involved in pain perception, pain processing, and pain management like opioid receptors, transporters, and other targets of pharmacotherapy are under investigation [141]. Genetic differences (e.g., G118 allele) may explain why some patients need higher opioid doses and the adverse effects profile may be modified by these mutations [142]. Some genes (e.g., fetal hemoglobin) are expressed much more in early life than in adults, and gene switching may mean a drug is effective at one age and not another.
In adults, gene testing may prove valuable for reducing adverse drug effects [143, 144]. However, most drug responses involve a large number of proteins regulated by multiple genes. Genotype does not equate with phenotype; environment, concomitant therapy, and disease have impact, and allele prevalence varies among ethnic groups [145]. The situation in children is more complex. Allelic variants may remain unchanged throughout life, but transcriptomic, proteomic, and metabolomic data in children are continuously changing throughout the development.
Pharmacodynamic Differences in the First Year of Life
Children’s responses to drugs have much in common with the responses in adults [146]. For example, acetaminophen analgesia appears similar in both neonates and children [147]. The perception that drug effects differ in children arises because the drugs have not been adequately studied in pediatric populations who have size- and maturation-related effects as well as different diseases. Neonates and infants, however, often do have altered pharmacodynamics.
The minimal alveolar concentration (MAC) for almost all anesthetic vapors is less in neonates than in infancy, which is in turn greater than that observed in children and adults [33]. MAC of isoflurane in preterm neonates less than 32 weeks gestation was 1.28 %, and MAC in neonates 32–37 weeks gestation was 1.41 % [148]. This value rose to 1.87 % by 6 months before decreasing again over childhood [148]. The cause of these differences is uncertain and may relate to maturation changes in cerebral blood flow, gamma-aminobutyric acid (GABAA) receptor numbers, or developmental shifts in the regulation of chloride transporters. Delivery of halothane to neonates at a MAC suitable for adults contributed to bradycardia and increased mortality in neonates [149].
The dose of thiopentone varies with age, e.g., 3.4 mg/kg in neonates, 6.3 mg/kg in infants, and 4.5 mg/kg in children 4–7 years [150, 151]. It remains uncertain whether altered pharmacokinetics or pharmacodynamic responses explain the reduced dose requirements in neonates. The effect site concentration of thiopentone for induction of anesthesia in neonates may be less than that in infants because the neonate has relatively immature cerebral cortical function, rudimentary dendritic arborizations, and relatively few synapses, but there are no studies to support or refute this premise.
Neonates have an increased sensitivity to the effects of NMBDs [121]. The reason for this is unknown but it is consistent with the observation that there is a threefold reduction in the release of acetylcholine from the infant rat phrenic nerve [152, 153]. The increased volume of distribution, however, means that a single NMBD dose is the same as the older child; reduced clearance prolongs duration.
Cardiac calcium stores in the endoplasmic reticulum are reduced in the neonatal heart because of immaturity. Exogenous calcium has greater impact on contractility in this age group than in older children or adults. Conversely, calcium channel blocking drugs (e.g., verapamil) can cause life-threatening bradycardia and hypotension [154]. There are some data to suggest greater sensitivity to warfarin in children, but the mechanism is not determined [155]. Amide local anesthetic agents induce shorter block duration and require a larger weight-scaled dose to achieve similar dermatomal levels when given by subarachnoid block to infants. This may be due, in part, to myelination, spacing of nodes of Ranvier, and length of nerve exposed as well as size factors and cerebrospinal fluid volume changes with age. There is an age-dependent expression of intestinal motilin receptors and the modulation of gastric antral contractions in neonates. Prokinetic agents may not be useful in very preterm infants, partially useful in older preterm infants, and useful in full-term infants. Similarly, bronchodilators in infants are ineffective because of the paucity of bronchial smooth muscle that can cause bronchospasm.
Measurement of PD Endpoints
Outcome measures are more difficult to assess in neonates and infants than in children or adults. Measurement techniques, disease and pathology differences, inhomogeneous groups, recruitment issues, ethical considerations, and endpoint definition for establishing efficacy and safety confuse data interpretation [156].
Common effects measured include anesthesia depth, pain, and sedation and neuromuscular blockade. A common effect measure used to assess depth of anesthesia is the electroencephalogram or a modification of detected EEG signals (spectral edge frequency, bispectral index [BIS], entropy). Physiological studies in adults and children indicate that EEG-derived anesthesia depth monitors can provide an imprecise and drug-dependent measure of arousal. Although the outputs from these monitors do not closely represent any true physiological entity, they can be used as guides for anesthesia and in so doing have improved outcomes in adults. In older children, the physiology, anatomy, and clinical observations indicate the performance of the monitors may be similar to that in adults. In infants their use cannot yet be supported in theory or in practice [157, 158]. During anesthesia, the EEG in infants is fundamentally different from the EEG in older children; there remains a need for specific neonate-derived algorithms if EEG-derived anesthesia depth monitors are to be used in neonates [159, 160].
New monitoring devices continue to be investigated. For example, a mid-latency auditory evoked potential-derived index of depth of hypnosis was recently investigated, but found inferior to BIS for differentiating different levels of sedation [161]. Alternative EEG signal-processing devices show promise and have been used to deliver closed-loop anesthesia [162].
The Children’s Hospital of Wisconsin Sedation Scale [163] has been used to investigate ketamine in the emergency department [164]. However, despite the use of such scales in procedural pain or sedation studies, few behavioral scales have been adequately validated in this setting [165, 166]. Interobserver variability can be high [167]. The COMFORT sedation scale [168] is one scoring system that is finding increasing usefulness in the pediatric intensive care setting. It is well validated [169], appears useful in different cultures, and has even been extended to premature neonates [170] and children with Trisomy 21 [171]. Changing patterns of drug use and reconsideration of what constitutes important adverse events may result in new definitions of the sedation continuum [172, 173]. Unfortunately, most pain scores are validated for the acute, procedural setting and perform less for subacute or chronic pain or stress. Postoperative nausea is difficult to quantify in neonates and infants who cannot verbalize; this makes comparison with adult postoperative nausea and vomiting scales tenuous.
Population Modeling
Mathematical models describe complex systems in simple terms, enabling us to describe, predict, and explain observations. Pharmacokinetic (PK) and pharmacodynamic (PD) models are used to improve pediatric anesthetic and sedation management. They quantify the exposure–response relationship, often providing clarity and insight into complex systems as well as a mechanistic understanding of the drug effect. Dose selection can be rationalized. Models may enable extrapolation beyond observed data. Modeling is a knowledge management tool; it captures and integrates data from all studies. Models can also be used for hypothesis testing and can drive decision-making during drug development.
Population PK and PD modeling using nonlinear mixed effects models has had enormous impact in adult anesthetic pharmacology. This methodology has particular applicability in children where the blood volume available for sampling is limited. Sparse data from multiple subjects can be used. Sampling times are not crucial for population methods and can be fitted around clinical procedures or outpatient appointments. Sampling time bands rather than exact times are equally effective and allow flexibility in neonates. Sampling cannulae used for PK studies may block or tissue, parents may refuse repeat sampling, and repeat venipuncture is frowned upon. Missing data, however, can still be used in a pediatric population analysis. Data from different studies can be pooled [174, 175].
The Target Concentration Approach
The goal of treatment is the target effect. (Refer to Chap. 31.) A pharmacodynamic model is used to predict the target concentration given a target effect. Population estimates for the PD model parameters and covariate information are used to predict typical PD values in a specific patient. Population estimates of PK model parameter estimates and covariate information are then used to predict typical PK values in a typical patient. For example, a dexmedetomidine steady-state target concentration of 0.6 μg/L may be achieved with an infusion of 0.33 μg/kg/h in a neonate, 0.51 μg/kg/h in a 1-year-old, and 0.47 μg/kg/h in an 8-year-old [109]. This target concentration strategy is a powerful tool for determining clinical dose [176]. Monitoring of serum drug concentrations and Bayesian forecasting may be used to improve dosing in individual patients.
This target effect approach is intrinsic to pediatric anesthetists using target-controlled infusion systems. These devices target a specific plasma or effect site concentration in a typical individual, and this concentration is assumed to have a typical target effect. The target concentration is one that achieves target therapeutic effect (e.g., anesthesia or a sedation level) without excessive adverse effects (e.g., hypotension). Effect monitoring (e.g., BIS) can be used to refine the target effect.
Pharmacokinetic Models
Compartment models dominate the sedative and analgesic literature. Standard compartment models may be unable to accurately describe drug concentrations immediately after bolus administration of an anesthetic induction agent because mixing in the central compartment is not instantaneous, making it difficult to model the fast blood-to-brain concentration equilibrium [177], and pulmonary uptake may also occur [178]. Recirculatory models help explain these early phase PK [179]. Such models have proved valuable determining anesthetic induction doses [180] and NMBD pharmacodynamics [181]. Physiologically based pharmacokinetic (PBPK) modeling has been used to assist with first-time dosing in children. A general PBPK model for drug disposition in infants and children, covering the age range from birth to adulthood, has been successfully evaluated using theophylline and midazolam as model drugs [60].
A single compartment is often insufficient to characterize the time-concentration profile, and further compartments are required (mammillary models). Drug is administered into a central compartment (V1) and redistributes to peripheral compartments (V2, V3, etc.; Fig. 11.4a). In a two-compartment model, transfer of drug between the central and peripheral compartment is relatively fast compared with the rate of elimination. Such models can be applied to both intravenous and inhalational drugs. The anesthetic conserving device (AnaConDa) delivering sevoflurane has also been described using compartment models [182]. A plot of the natural log of concentration after bolus reveals two distinct slopes (rate constants, α[alpha] and β[beta], Fig. 11.4b). Consequently the time-concentration profile is commonly described using a polyexponential function:
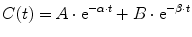 These polyexponential parameters have little connection with underlying physiology, and an alternative parameterization is the use of a central volume and three rate constants (k 10, k 12, k 21) that describe drug distribution between compartments. Another common method is to use two volumes (V 1, V 2) and two clearances (CL, Q). Q is the inter-compartment clearance.
These polyexponential parameters have little connection with underlying physiology, and an alternative parameterization is the use of a central volume and three rate constants (k 10, k 12, k 21) that describe drug distribution between compartments. Another common method is to use two volumes (V 1, V 2) and two clearances (CL, Q). Q is the inter-compartment clearance.
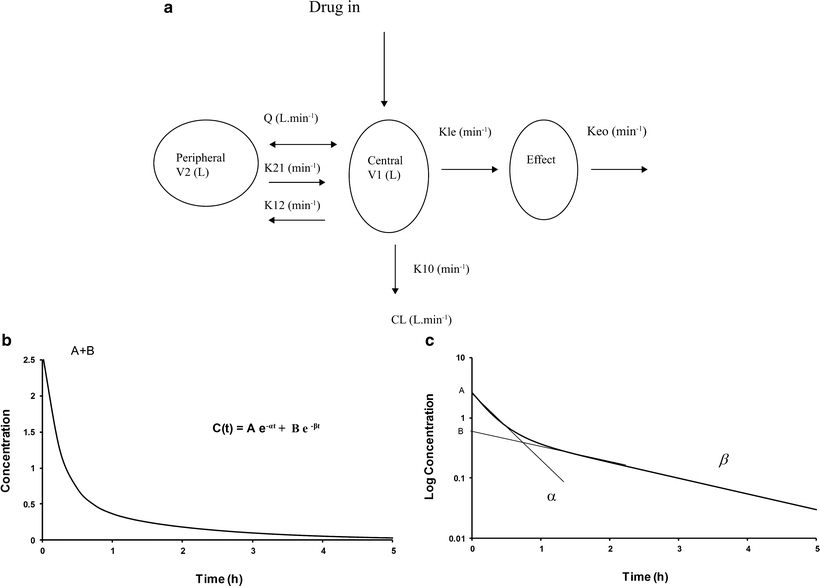
Fig. 11.4
(a) A mammillary two-compartment PK model with additional compartment for effect. (Reprinted with permission from Anderson BJ. Pediatric models for adult target-controlled infusion pumps. Paediatr Anaesth 2010 Mar;20(3):223-32.) (b) Time-concentration profile for a two-compartment model. (c) Conversion of concentration to a log scale allows estimation of elimination constants and compartment volumes
Students are commonly taught to estimate compartment model PK parameters through interpretation of graphs representing time-concentration profiles. Conversion of concentration to a log scale allows estimation of elimination constants and compartment volumes (Fig. 11.4c). Integration of the function describing this profile yields an AUC (area under the curve), from which CL can be determined:
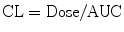 Computers have made the use of nonlinear regression to directly estimate parameters through iterative techniques using least squares curve fitting. Models with two or more compartments are now commonly solved using differential equations.
Computers have made the use of nonlinear regression to directly estimate parameters through iterative techniques using least squares curve fitting. Models with two or more compartments are now commonly solved using differential equations.
Parameter estimates (CL, Q, V 1, V 2) can be used to predict dose. A loading dose raises concentration in the plasma to target concentration promptly and may be desirable in anesthesia when rapid effect is required. In a one-compartment model, the volume of distribution is the proportionality factor that relates total amount of drug in the body to plasma concentration (TC = target concentration):
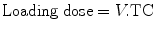 This calculation may not be applicable to many sedative drugs that are characterized using multi-compartment models. The use of V 1 results in a loading dose too high; too high a dose may cause transient toxicity.
This calculation may not be applicable to many sedative drugs that are characterized using multi-compartment models. The use of V 1 results in a loading dose too high; too high a dose may cause transient toxicity.
An alternative technique is to use the target effect dose. The time to peak effect (T peak) is dependent on clearance and effect site equilibration half-time (T 1/2keo). At a submaximal dose, T peak is independent of dose. At supramaximal doses, maximal effect will occur earlier than T peak and persist for longer duration. The T peak concept has been used to calculate optimal initial bolus doses [183].
Clearance is the most important parameter when defining a rational steady-state dosage regimen. At steady state:
Dosing ratess = rate of eliminationss = CL ⋅ TC
When a drug is given intermittently:
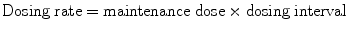 When a drug is given by constant infusion:
When a drug is given by constant infusion:
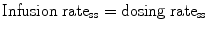 Once the target concentration of a drug is defined, the infusion rate is determined by CL at steady state. Many sedative drugs distribute to peripheral compartments and steady state may not be achieved during the time of infusion. Dose adjustment is required to achieve constant effect until steady-state conditions are reached.
Once the target concentration of a drug is defined, the infusion rate is determined by CL at steady state. Many sedative drugs distribute to peripheral compartments and steady state may not be achieved during the time of infusion. Dose adjustment is required to achieve constant effect until steady-state conditions are reached.
Propofol PK are usually described using a three-compartment mammillary model. In order to achieve steady state, 3 μg/mL in children 3–11 years, dosing changes are required, e.g., a loading dose of 2.5 mg/kg followed by an infusion rate of 15 mg/kg/h for the first 15 min, 13 mg/kg/h from 15 to 30 min, 11 mg/kg/h from 30 to 60 min, 10 mg/kg/h from 1 to 2 h, and 9 mg/kg/h from 2 to 4 h. Target-controlled infusion (TCI) pumps are capable of fine-tuning by making adjustments at 10 s intervals [184].
The PK of drug disposition confined to a one-compartment model is often expressed in terms of half-life. Half-life (T 1/2) is the time required to change the amount of drug in a body compartment by one-half:
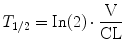 This half-life is related to the elimination rate constant (k), a parameter representing the slope of the exponential decay curve:
This half-life is related to the elimination rate constant (k), a parameter representing the slope of the exponential decay curve:
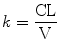 Elimination half-life is of no value in characterizing disposition of intravenous anesthetic drugs with multiple compartments during dosing periods relevant to anesthesia. A more useful concept is that of the context-sensitive half-time where “context” refers to infusion duration. This is the time required for the plasma drug concentration to decline by 50 % after terminating infusion [185]. The context-sensitive half-time is the same as the elimination half-life for a one-compartment model and does not change with infusion duration.
Elimination half-life is of no value in characterizing disposition of intravenous anesthetic drugs with multiple compartments during dosing periods relevant to anesthesia. A more useful concept is that of the context-sensitive half-time where “context” refers to infusion duration. This is the time required for the plasma drug concentration to decline by 50 % after terminating infusion [185]. The context-sensitive half-time is the same as the elimination half-life for a one-compartment model and does not change with infusion duration.
Context-sensitive half-time may be independent of infusion duration (e.g., remifentanil 2.5 min), moderately affected (propofol 12 min at 1 h, 38 min at 8 h), or display marked prolongation (e.g., fentanyl 1 h at 24 min, 8 h at 280 min). This is due to return of drug to plasma from peripheral compartments after ceasing infusion. Peripheral compartment size differs in children from adults, so that at termination of infusion, more drug may remain in the body for any given plasma concentration than in adults. The context-sensitive half-time for children given propofol, for example, is longer [184]. The context-sensitive half-time gives insight into PK of a hypnotic drug, but the parameter may not be clinically relevant because the percentage decrease in concentration required for recovery is not necessarily 50 %.
Pharmacodynamic Models
Pharmacokinetics is what the body does to the drug, while pharmacodynamics is what the drug does to the body. The precise boundary between these two processes is ill defined and often requires a link describing movement of drug from the plasma to the effect site and its target. Drugs may exert effect at nonspecific membrane sites, by interference with transport mechanisms, by enzyme inhibition or induction, or by activation or inhibition of receptors.
The Sigmoid E max Model
The relation between drug concentration and effect may be described by the Hill equation (see maturation model above), well known to anesthesiologists through the oxygen dissociation curve [84], according to the equation:
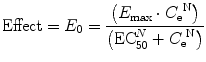 where E 0 is the baseline response, E max is the maximum effect change, C e is the concentration in the effect compartment, EC50 is the concentration producing 50 % E max, and N is the Hill coefficient defining the steepness of the concentration–response curve. Efficacy is the maximum response on a dose or concentration–response curve. EC50 can be considered a measure of potency relative to another drug, provided N and E max for the two drugs are the same. A concentration–response relationship for acetaminophen has been described using this model. An EC50 of 9.8 mg/L, N = 1, and an E max of 5.3 pain units (VAS 0–10) was reported [186]. Midazolam PD in adults has been similarly defined using EEG response [187, 188]. Adverse effects can also be described using this model; the reduced cardiac output observed as dexmedetomidine concentration increases has been expressed using a sigmoid E max model with a EC50 of 2.4 μg/L and N 3.15 [117]. Blood pressure changes in children given dexmedetomidine have also been described using Emax models [189].
where E 0 is the baseline response, E max is the maximum effect change, C e is the concentration in the effect compartment, EC50 is the concentration producing 50 % E max, and N is the Hill coefficient defining the steepness of the concentration–response curve. Efficacy is the maximum response on a dose or concentration–response curve. EC50 can be considered a measure of potency relative to another drug, provided N and E max for the two drugs are the same. A concentration–response relationship for acetaminophen has been described using this model. An EC50 of 9.8 mg/L, N = 1, and an E max of 5.3 pain units (VAS 0–10) was reported [186]. Midazolam PD in adults has been similarly defined using EEG response [187, 188]. Adverse effects can also be described using this model; the reduced cardiac output observed as dexmedetomidine concentration increases has been expressed using a sigmoid E max model with a EC50 of 2.4 μg/L and N 3.15 [117]. Blood pressure changes in children given dexmedetomidine have also been described using Emax models [189].
Quantal Effect Model
The potency of anesthetic vapors may be expressed by MAC (minimum alveolar concentration), and this is the concentration at which 50 % of subjects move in response to a standard surgical stimulus. MAC appears at first sight to be similar to EC50, but is an expression of quantal response rather than magnitude of effect. There are two methods of estimating MAC. Responses can be recorded over the clinical dose range in a large number of subjects and logistic regression applied to estimate the relationship between dose and quantal effect; the MAC can then be interpolated. Large numbers of subjects may not be available and so an alternative is often used. The “up-and-down” method described by Dixon [190, 191] estimates only the MAC rather than the entire sigmoid curve. It involves a study of only one concentration in each subject and, in a sequence of subjects, each receives a concentration depending upon the response of the previous subject; the concentration is either increased if the previous subject did not respond or decreased if they did. The MAC is usually calculated either as the mean concentration of equal numbers of responses and no-responses or is the mean concentration of pairs of “response-no response.” This method has also been applied to drugs other than inhalation vapors, e.g., local anesthesia dose for spinal block [192, 193].
Logistic Regression Model
When the pharmacological effect is difficult to grade, then it may be useful to estimate the probability of achieving the effect as a function of plasma concentration. Effect measures such as movement/no movement or rousable/non-rousable are dichotomous. Logistic regression is commonly used to analyze such data, and the interpolated EC50 value refers to the probability of response. For example, an EC50 of 0.52 mg/L for arousal after ketamine sedation in children has been estimated using this technique [164].
Linking PK with PD
A simple situation in which drug effect is directly related to concentration does not mean that drug effects parallel the time course of concentration. This occurs only when the concentration is low in relation to EC50. In this situation, the half-life of the drug may correlate closely with the half-life of drug effect. Observed effects may not be directly related to serum concentration. Many drugs have a short half-life but a long duration of effect. This may be attributable to induced physiological changes (e.g., aspirin and platelet function) or may be due to the shape of the E max model. If the initial concentration is very high in relation to the EC50, then drug concentrations 5 half-lives later, when we might expect minimal concentration, may still exert considerable effect [74]. There may be a delay due to transfer of the drug to effect site (NMBD), a lag time (diuretics), physiological response (antipyresis), active metabolite (propacetamol), or synthesis of physiological substances (warfarin).
A plasma concentration–effect plot can form a hysteresis loop because of this delay in effect. Hull [194] and Sheiner [195] introduced the effect compartment concept for muscle relaxants. The effect compartment concentration is not the same as the blood or serum concentration and is not a real measurable concentration. It has negligible volume and contains negligible blood. A single first order parameter (T 1/2keo) describes the equilibration half-time. This mathematical trick assumes concentration in the central compartment is the same as that in the effect compartment at equilibration, but that a time delay exists before drug reaches the effect compartment. The concentration in the effect compartment is used to describe the concentration–effect relationship [196].
Adult T 1/2keo values are well described, e.g., morphine 16 min, fentanyl 5 min, alfentanil 1 min, and propofol 3 min. This T 1/2keo parameter is commonly incorporated into TCI pumps in order to achieve a rapid effect site concentration. The adult midazolam T 1/2keo of 5 min [197] may be prolonged in the elderly [198, 199], resulting in overdose if this is not recognized during dose titration.
Onset of drug effect is quicker with decreasing age. We might expect a shorter propofol T 1/2keo with decreasing age based on size models [200], and this is exactly what has been described by Jeleazcov et al. [201]. Similar results have been demonstrated for sevoflurane and BIS [202]. If unrecognized, this will result in excessive dose in a young child if the effect site is targeted and peak effect (T peak) is anticipated to be later than it actually is because it was determined in a teenager or adult. Unfortunately, integrated PKPD studies in children are lacking. Available pediatric propofol T 1/2keo values have been determined by application of published PK data to PD observations only [203–205].
Adverse Effects
Neonates and young children may suffer permanent effects resulting from a stimulus applied at a sensitive point in development. For example, congenital hypothyroidism, if untreated, causes lifelong phenotypic changes. The incidence of vaginal carcinoma is high in children of mothers treated with stilboestrol during pregnancy [206]. There are concerns that neonatal exposure to some anesthetic agents (e.g., ketamine, midazolam) may cause widespread neuronal apoptosis and long-term memory deficits [207, 208].
Anesthesia, analgesia, or sedation generally involves examination of immediate adverse effects such as PONV, hypotension, or respiratory depression. A dose–response curve for intravenous morphine and vomiting was investigated in children having day-stay tonsillectomy. Doses above 0.1 mg/kg were associated with a greater than 50 % incidence of vomiting [209]. These data are similar to those in children undergoing inguinal herniorrhaphy [210], suggesting that lower doses of morphine are associated with a decreased incidence of emesis after day-stay surgery, and encourage the use of alternative analgesic drugs.
Therapeutic use of drugs balances beneficial effects against adverse effects. Adverse effects, however, may be simply consequent upon a poor understanding of pharmacokinetics. Propofol infusion dose in neonates, if based on adult dose (mg/kg/h), will overdose and cause hypotension; propofol infusion dose in 1–2-year-olds (where clearance is increased expressed as mg/kg/h) may underdose and result in awareness. Morphine dose in the very young was traditionally limited by fears of respiratory compromise; postoperative arterial oxygen desaturation continues to be reported with sedative drugs in neonates [211]. These are a result of poor pharmacokinetic understanding. However, there are also pharmacodynamic differences. Premature neonates are more prone to apnea. Sympathetic–parasympathetic tone is immature in neonates, and the use of propofol in neonates has recently been associated with profound hypotension [212], questioning our understanding of the dose–effect relationships of this common drug [213]. Such information allows informed dosing.
Drug Interactions
Drug interactions can increase or decrease response mediated through either PK or PD routes. Phenobarbitone induces a number of other pathways responsible for drug clearances, e.g., CYP1A2, CYP2C9, CYP2C19, CYP3A4, and UDP-glucuronosyltransferase (UGT) [214]. Ketamine in humans is metabolized mainly by CYP3A4. The steep concentration–response curve described for ketamine [164] means that small changes in the plasma concentration attributed to increased clearance can have dramatic impact on the degree of sedation [215].
An increase in the T 1/2keo of d-tubocurarine with increasing inspired halothane concentrations has been demonstrated [216]. Halothane is a negative inotrope [217] and reduces skeletal muscle blood flow [218], so it seems reasonable to interpret changes in T 1/2keo as due to changes in blood flow. Inhalation anesthetic agents can also prolong duration of block and this effect is agent specific. Sevoflurane potentiated vecuronium more than halothane; when compared to balanced anesthesia, the dose requirements of vecuronium were reduced by approximately 60 % and 40 %, respectively [219].
Anesthetic drug interactions traditionally have been characterized using isobolographic analysis or multiple logistic regression. Minto et al. [220] have proposed a model based on response surface methodology. Computer simulations based on interactions at the effect site predicted that the maximally synergistic three-drug combination (midazolam, propofol, and alfentanil) tripled the duration of effect compared with propofol alone. Response surfaces can describe anesthetic interactions, even those between agonists, partial agonists, competitive antagonists, and inverse agonists [220].
Synergism between propofol and alfentanil has been demonstrated using response surface methodology. Remifentanil alone had no appreciable effect on response to shaking and shouting or response to laryngoscopy while propofol could ablate both responses. Modest remifentanil concentrations dramatically reduced the concentrations of propofol required to ablate both responses [221]. When comparing the different combinations of midazolam, propofol, and alfentanil, the responses varied markedly at each endpoint assessed and could not be predicted from the responses of the individual agents [222]. Similar response surface methodology has been taken for investigation of the combined administration of sevoflurane and alfentanil [223] and remifentanil and propofol [224] on ventilation control. These combinations have a strikingly synergistic effect on respiration, resulting in severe respiratory depression in adults. These synergistic associations can be extended to pediatric sedation techniques. It is little wonder that the use of three or more sedating medications compared with one or two medications was strongly associated with adverse outcomes [7].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


