Fig. 11.1
Panoramic radiograph of a 31-year-old male patient with giant cell tumors of the right maxilla and mandible (arrows). The lesions are associated with root resorption of the adjacent teeth
Based on biologic behavior, GCTs are classified into two groups, as originally described by Chuong and Kaban in 1985 [3]. Aggressive lesions are ≥5 cm in size, display recurrence after treatment, or have three of the following characteristics: root resorption, tooth displacement, cortical bone thinning, or cortical perforation. The more common non-aggressive lesions lack these criteria and are typically asymptomatic. There is some evidence to suggest that the difference in biologic behavior may be related to increased levels of angiogenic factors in aggressive lesions [5, 6].
Treatment protocols are based on biologic behavior. Non-aggressive lesions predictably respond to enucleation/curettage. Aggressive lesions are treated with en bloc resection to achieve histologically clear margins (1 cm), or with nerve/tooth-sparing enucleation with adjuvant anti-angiogenic therapy (such as interferon-alpha, IFN) [7–9]. Patients who have difficulty with interferon therapy (lethargy, malaise, fevers, etc.) can be successfully managed with bisphosphonate therapy. However, bisphosphonates should be used with caution if dental extractions are anticipated, due to the potential risk of osteonecrosis [1, 2].
Cherubism
Cherubism is a rare autosomal dominant condition resulting from mutation in the SH3BP2 gene (4p16.3) [10–18]. Cherubism is characterized by bilateral, symmetric GCTs in the facial bones most commonly in the mandible. Maxillary lesions and extragnathic involvement (ribs, humerus) have been reported [10, 15–17]. The condition is typically diagnosed around ages 7–10 with initial presentation around 2–5 years of age [10]. Replacement of mandibular bone with fibrous tissue gives rise to the classic appearance of “cherubic” faces, originally described by Dr. William A. Jones in 1933 [11, 12]. Involvement of the bones of the orbit can result in an upward gaze (“eyes towards heaven”). The presence of these lesions may be associated with premature loss of primary teeth and delayed eruption of permanent teeth. Extensive bony involvement can result in vision and hearing changes. In most cases, the condition becomes quiescent during late adolescence, with the facial features of most adult patients approaching normalcy by age 40 [13, 14].
Radiographically, the lesions appear as bilateral symmetric multilocular radiolucencies with associated expansion of the bony cortices (Fig. 11.2). Treatment consists of observation. Once the lesions become quiescent, treatment is aimed at recontouring excessive fibrous tissue to reestablish normal bony contours. Early, aggressive intervention has been associated with regrowth and worsening deformity. Radiation therapy is contraindicated due to reports of sarcomatous degeneration [10]. During active clinical growth, annual plain film imaging is recommended. Following established quiescence, plain film imaging is recommended every 3–5 years [10, 14].
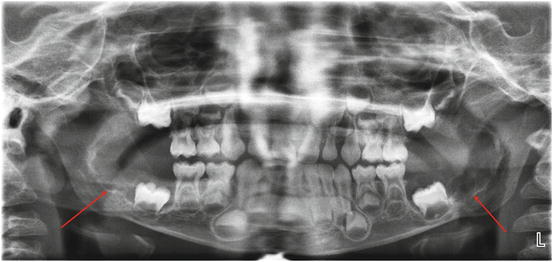
Fig. 11.2
Panoramic radiograph of a 4-year-old male with cherubism. Bilateral multilocular radiolucencies are seen in the posterior mandible (arrows)
Noonan/Multiple Giant Cell Lesion Syndrome
Noonan syndrome was first described in 1963 by Dr. Jaqueline Noonan at the University of Iowa. Patients with the classic phenotype have short stature, hypertelorism, posteriorly angulated and low-set ears, congenital cardiac defects (most commonly pulmonic valve stenosis), bleeding diatheses (von Willebrand disease, factor deficiencies, thrombocytopenia), developmental delay, and cryptorchidism in males [19, 20]. In 1991, Cohen and Gorlin identified 15 cases of patients with Noonan syndrome and giant cell lesions of the jaws [21]. They coined the term Noonan/multiple giant cell lesion syndrome (NMGCLS) and considered it to be a distinct entity from Noonan syndrome and cherubism.
More recently, mutations in the SOS1 and PTPN11 genes have been identified in patients with Noonan syndrome, allowing for a molecular distinction between Noonan syndrome and cherubism [22, 23]. Molecular analyses have provided evidence to suggest that NMGCLS is a variant along the spectrum of Noonan syndrome [24, 25].
It is important for the clinician to distinguish between NMGCLS and cherubism. Clinical behavior of the associated GCTs, and thus, treatment, can be dramatically different. In NMGCLS, GCTs can demonstrate aggressive behavior and lead to significant damage to surrounding tissues [10, 26]. Genetic testing becomes particularly important in cases where there is bilateral, symmetric jaw involvement, as it may be difficult to distinguish cherubism from NMGCLS clinically or radiographically (Fig. 11.3).
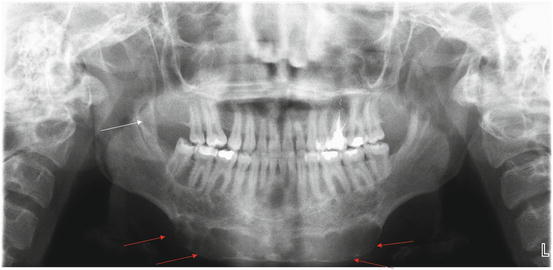
Fig. 11.3
Panoramic radiograph of a 19-year-old female with Noonan multiple giant cell lesion syndrome. An expansile, multilocular radiolucency is seen in the anterior mandible (red arrows). A smaller lesion is evident in the right mandibular ascending ramus (white arrow) (Radiograph courtesy of Leonard B. Kaban, DMD, MD)
Differentiation between the GCTs in NMGCLS and cherubism has important consequences for management. Typically, GCTs in Noonan syndrome behave more aggressively and usually require surgical intervention with adjuvant treatment [10]. Thus, it is imperative that the practitioner utilize clinical exam and diagnostic testing to differentiate between the different entities.
Fibrous Dysplasia
In fibrous dysplasia (FD), fibrous tissue and woven bone replace normal bone and marrow [27, 28]. FD is caused by somatic activating mutations in the a-subunit of the Gs protein of GNAS gene [29, 30]. Monostotic FD is more common; polyostotic FD may include McCune–Albright syndrome (polyostotic fibrous dysplasia, café-au-lait macules, precocious puberty, endocrinopathy). Patients are typically between ages 5 and 20 [27]. Clinically, patients may be asymptomatic or present with a painless swelling. Growth of the lesions tends to correlate with somatic growth and hormonal changes, with the fastest rates observed during childhood and adolescence. Quiescence typically occurs by age 25. Recrudescent growth may be seen during periods of hormonal changes (e.g., pregnancy, use of oral contraceptives). Major growth changes outside of these events are unusual and should alert the clinician to pursue additional workup.
In the maxillofacial skeleton, the most common sites of involvement are the maxilla and the mandible. In the maxilla, involvement of the orbital floor may produce paresthesia or proptosis [27]. Mandibular FD is most common at the angle; it must be distinguished from masseter hypertrophy, primary jaw tumors, or osteomyelitis. Radiographically, FD appears as multilocular radiolucencies or mixed radiolucent–radiopaque appearance (Fig. 11.4). On CT, FD has characteristic “ground glass” appearance (Fig. 11.5) [28]. Delayed dental eruption and splaying of the roots in the affected regions are frequently seen (Fig. 11.6). Serum levels of calcium, phosphate, and alkaline phosphatase are typically normal for monostotic fibrous dysplasia. In patients with McCune–Albright syndrome, disease activity can be monitored with serum alkaline phosphatase levels [31].
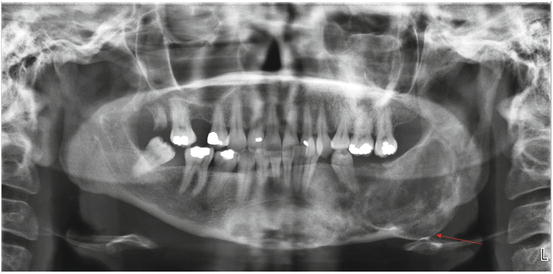
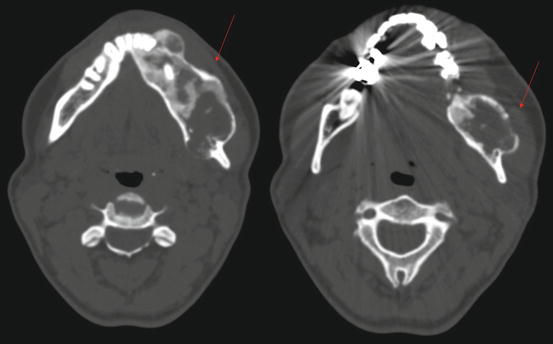
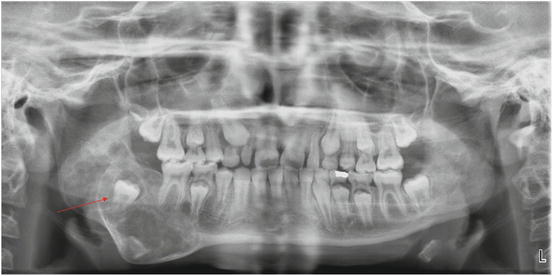
Fig. 11.4
Panoramic radiograph of a 44-year-old female with fibrous dysplasia of the mandible. Multilocular, mixed lesions are seen in the left mandibular body and mandibular symphysis, with associated cortical expansion (arrows) (Radiograph courtesy of Leonard B. Kaban, DMD, MD)
Fig. 11.5
Maxillofacial axial computed tomography of the patient from Fig. 11.4. Notice classic “ground glass” appearance and cortical expansion (arrows)
Fig. 11.6
Panoramic radiograph of an 11-year-old female with fibrous dysplasia of the right mandible. There is a large, expansile, multilocular radiolucency in the right posterior mandible, with displacement of the right mandibular third molar (arrow) (Radiograph courtesy of Leonard B. Kaban, DMD, MD)
Management of FD is twofold [8]. First, biopsy is done to confirm the diagnosis. Asymptomatic, quiescent, or slow growing FD can be enucleated, observed, or contoured. Patients with significant asymmetry, paresthesia, and/or proptosis are considered to have aggressive/rapidly growing FD. Management consists of symptomatic debulking/contour resection [27]. Among patients with quiescent disease, rapid growth, new onset of pain, paresthesia, and/or functional deficit (e.g., hearing or vision changes, trismus, nasal obstruction) can indicate disease reactivation and surgical intervention should be considered. Among adult patients, contouring can be performed after 1 year of documented clinical stability. Similar to cherubism, sarcomatous change has been reported following radiotherapy; such treatment should be avoided [2, 10].
Vascular Malformations
Vascular malformations occur as a result of aberrant morphogenesis of blood vessels or lymphatic structures [32–35]. Vascular malformations are present at birth but may not become clinically apparent until late infancy or childhood [33, 34]. Unlike vascular tumors, vascular malformations grow proportionately with the patient and do not regress. They may increase in size at any time including somatic growth, trauma, infection, or hormonal changes such as puberty and pregnancy [32–36]. Vascular malformations are categorized according to the vessel type involved (capillary, lymphatic, venous, arterial) and the rate of flow (slow or fast) [32–35]. While most are composed of a single-type of vascular entity, combined malformations have been described such as capillary-lymphatic, capillary-venous, lymphatico-venous, and capillary lymphatico-venous [34–36]. The biology of vascular malformations has been studied extensively. Distinction between a vascular malformation and a vascular tumor must be made. Vascular malformations enlarge by dilatation of abnormal vasculature, whereas vascular tumors demonstrate proliferative growth. As such, vascular malformations are typically not treated by resection, with the exception of some low-flow venous malformations (VMs) of the jaws.
The clinical presentations of vascular malformations vary according to the type of vessel involved. Capillary malformations (CMs) change from pink, with a smooth surface in infancy, darkening through childhood, and appearing purple, with a tessellated surface in older patients. Venous malformations (VMs) are typically soft, easily compressible, bluish-hued lesions that may have associated palpable masses (phleboliths). They may be present on the skin or mucosa, or may span multiple tissue planes. Their size typically changes with maneuvers that increase venous pressure (dependency or Valsalva) [35]. Lymphatic malformations (LMs) are typically colorless while lymphatic-venous malformations are blue-purple. LMs can expand secondary to intralesional bleeding, bacterial infection, or during a period of lymphatic “stress” (e.g., upper respiratory infections). LMs with large cystic cavities (historically referred to as “cystic hygromas”) can be identified with transillumination. Both lymphatic and capillary-lymphatic malformations have irregular surfaces [34, 35].
Arteriovenous malformations (AVMs) are high flow lesions and are typically warm and tender to palpation. In cases where there is shunting, pulsatile flow and a palpable bruit may be present. Intraoral AVMs cause gingival hypertrophy, mucosal staining, and intraoral gingival bleeding. Teeth in the vicinity may be periodontally compromised and may demonstrate gross mobility [36–38].
In approximately 30 % of cases, vascular malformations are associated with skeletal changes such as difference in morphology or bone density adjacent to the malformation, or a frank intraosseous malformation [33–36]. The most common associated skeletal anomaly is overgrowth or expansion of bone deep to a slow-flow malformation [33–36]. The evolution of these changes has been difficult to elucidate, due to a paucity of radiographs taken at birth. However, it has been reported that approximately 80 % of patients with LM demonstrate evidence of altered skeletal growth by 10 years of age [35, 36].
Management of vascular malformations depends upon the vessel type and rate of flow. CMs typically do not require special precautions; tooth extractions, orthodontic treatment, and osteotomies can all be safely performed. LMs of the maxillofacial region typically involve the floor of mouth, mandible, and submandibular/cervical tissues. As such, feeding difficulties and airway obstruction become sources of considerable morbidity. These patients are typically managed with serial-staged excisions. Orthognathic surgery can take place once skeletal growth is completed.
Patients with venous or lymphatic-venous malformations who have dental or dentoskeletal deformities but no concomitant intraosseous involvement can have orthodontic treatment and orthognathic surgery without fear of excessive bleeding. Intraosseous involvement (i.e., microshunting) is associated with a higher risk for major hemorrhage, either from the malformation or from local or systemic coagulopathy [35]. In the latter situations, stasis and turbulence within the malformation lead to localized and, occasionally, disseminated intravascular coagulation. Prothrombin and partial thromboplastin times may be normal, but fibrin split products may be elevated and fibrinogen and platelet levels are reduced [35, 36]. Surgical intervention can take place once the coagulopathy is corrected; in the interim, patients are typically systemically anti-coagulated [35]. In cases of large venous malformations, direct injection of 100 % ethanol to sclerose the area has been demonstrated to be effective [35]. Smaller malformations may be sclerosed with injection of 1 % sodium tetradecyl sulfate [35, 36]. Tender phleboliths may be effectively treated with aspirin (325 mg daily) for an indefinite course [35].
High flow lesions, such as AVMs, are currently managed by staged procedures. The first stage involves occlusion of the nidus by arterial embolization. Proximal embolization and ligation are contraindicated due to the rapid formation of collateral flow. In the second stage (24–72 h post-embolization), surgical resection is completed, with the goal of complete resection. Unfortunately, AVMs are typically not well localized and the recurrence rates may be high (approximately 40 %) [39]. In recurrent cases, reoperation is not always feasible and repeat embolization is palliative [35, 39].
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) refers to a group of heritable disorders related to defects in collagen maturation. Both autosomal dominant and recessive patterns have been described. Most cases are associated with mutations in two genes involved in the formation of type I collagen: COL1A1 (Chromosome17) and COL1A2 (Chromosome 7) [40]. Due to the ubiquitous nature of type I collagen as a constituent of multiple types of connective tissues (bone, dentin, ligament, skin, etc.), the clinical manifestations vary. Abnormalities in collagen formation result in diffuse osteoporosis, bone with thin cortices, and aberrant callus formation with injury. In addition, affected individuals may have blue sclera, hearing loss, joint hyperextensibility, and alterations in tooth structure.
There is no definitive treatment for OI. Patients with OI are prone to fractures and aberrant healing and management can be complicated. Severe attrition of tooth structure and premature tooth loss are common. Surgical rehabilitation with osseointegrated implants is challenging due to the poor quality of bone. Among patients with skeletal malocclusion, orthognathic surgery can be performed, albeit with judicious planning [41–43].
Systemic Conditions Affecting Maxillofacial Structures
Patients with significant hematologic, renal, oncologic, or immunosuppressive conditions can be challenged with maxillofacial problems ranging from dental caries to osteonecrosis. These patients, while complicated from a medical treatment standpoint, do not necessarily require complex regimens for management of dental and maxillofacial conditions. In general, a patient should be medically optimized to the extent possible prior to elective surgical interventions. In situations where the risks of surgical interventions (e.g. sepsis, endocarditis, risks from anesthesia) outweigh the benefits, improvement of overall health and non-invasive intervention to improve oral health are performed. Fortunately, common surgical procedures in and around the oral cavity carry low attendant risks and, in most instances, can be carried out with minimal modifications to patient treatment protocols.
Treatment
Pharmacological therapies for conditions involving the pediatric maxillofacial skeleton are limited. No drugs are currently approved for the prevention of low bone density and possible pathologic fractures in children. However, therapies are currently in use off-label in clinical practice.
Vitamin D and Calcium Supplementation
Vitamin D deficiency is extremely common in the pediatric population [44, 45]. Although the level of vitamin D needed for bone health remains somewhat controversial, it has been shown that levels of 25-hydroxy vitamin D below 20 ng/mL increase bone turnover and the risk of low bone density [46]. In patients with conditions causing low bone density such as OI, supplementation to ensure a higher level of 25-hydroxy vitamin D of 30 ng/mL may be appropriate [47]. Evidence of the benefits of higher levels of supplementation on organ systems besides the skeleton is mixed [48]. There are few risks to vitamin D supplementation such as hypervitaminosis D and resulting hypercalcemia. Although rare, they have been reported at doses needed to maintain the above levels, indicating the need for monitoring of patients on supplementation [49].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


