Yogen Singh, Satyan Lakshminrusimha Acute pulmonary hypertension (aPH) in the neonatal period is common in sick newborn infants being treated in the neonatal intensive care unit (NICU). The causes and underlying pathophysiology are multifactorial. Acute pulmonary hypertension in the newborn is secondary to impaired or delayed relaxation of the pulmonary vasculature associated with a diverse group of cardiopulmonary pathologies such as meconium aspiration syndrome (MAS), congenital diaphragmatic hernia (CDH), sepsis, respiratory distress syndrome (RDS), prolonged premature rupture of membranes (PPROM), persistent hypoxia, hypoxic-ischemic encephalopathy (HIE), congenital heart disease CHD), heart failure, and alveolar capillary dysplasia, or without a known cause (idiopathic).1,2 Whatever the cause, most infants with aPH present soon after birth. Hence often this clinical presentation is termed persistent pulmonary hypertension of the newborn (PPHN), previously referred to as persistent fetal circulation (PFC).3 Acute pulmonary hypertension (PH) is the phenotypical presentation of various underlying conditions – it is a syndrome of impaired circulatory adaptation at birth.4 The hallmark of aPH physiology is a sustained elevation of pulmonary vascular resistance (PVR) and labile hypoxemia after birth.5 Despite advances in understanding of perinatal pathophysiology and neonatal management strategies, its prevalence (2 per 1000 live births) has not changed significantly.1,6 Most infants with aPH are born at term or late preterm, although around 2% of cases are born prematurely.7 Acute PH remains one of the leading causes of critical illness in the NICU, with a mortality rate of 5–10%, and this has not changed significantly despite advances in management and better understanding of pathophysiology.1 It is critical to understand the etiopathogenesis, altered physiology, and impact of treatment interventions on the underlying pathophysiology, allowing adoption of a physiology-based approach which may help in decreasing morbidity and mortality. This chapter is primarily focused on pathophysiology and evaluation of aPH, including the role of advanced hemodynamic evaluation in assessing severity and management of aPH and controversies / clinical dilemmas around cardio-centric management. The pharmacological treatment of aPH (Chapter 5), chronic pulmonary hypertension, and PH associated with bronchopulmonary dysplasia (BPD) in premature infants (Chapter 26) and CDH in term infants (Chapter 29) have been described in other chapters. During the sixth World Symposium of Pulmonary Arterial Hypertension (PAH) held in 2018 in Nice, France, the classification of PAH was updated.8,9 Due to its particular anatomic and physiologic nature, aPH in the neonatal period has been moved to a separate subcategory – “Persistent PH of the newborn syndrome”. However, in our opinion controversy persists in its nomenclature and classification because aPH is neither “persistent” nor present from birth in all infants. It develops secondary to high PVR (which may fail to drop or worsen from an underlying condition) and right and left ventricular dysfunction leading to circulatory failure, associated with acute hypoxia. While the sixth World Symposium of PAH classification of pediatric pulmonary hypertension is useful, a more commonly used classification of aPH based upon etiology, primary (idiopathic) and secondary PPHN, would be more useful to the clinician (Figure 25.1). Compared to pediatric pulmonary hypertension, only around 10–20% of cases of aPH are idiopathic; most cases (80–90%) occur from abnormally constricted pulmonary vasculature due to MAS, sepsis, CDH, RDS, pneumonia, HIE / perinatal hypoxia, or CDH – this is referred to as secondary aPH. Recent data from California suggest that infection (30%), MAS (24%), idiopathic (20%), RDS (7%), and CDH (6%) are the five leading causes of aPH in newborns.1 In these cases it can be difficult to separate chronic intrauterine remodeling from acute pulmonary vasoconstriction due to parenchymal lung disease. Primary or idiopathic aPH refers to the absence of parenchymal lung disease/other neonatal disease to explain elevated pulmonary arterial pressure and implies intrauterine pulmonary vascular remodeling. In neonates this accounts for only 10–20% of cases. Some cases of idiopathic aPH may potentially have a genetic basis. The PVR during the fetal period is high due to mechanical (fluid-filled alveoli, prominent endothelium), biochemical (high levels of endothelin and serotonin and low levels of nitric oxide and prostacyclin – see Figure 25.2), and alveolar hypoxia.10,11 Chronic intrauterine hypoxia, exposure to non-steroidal anti-inflammatory drugs (NSAIDs),12,13 and serotonin-reuptake inhibitors (SSRI) have been associated with vascular remodeling and aPH. However, the association between NSAID use during pregnancy and aPH has been questioned.14 The imbalance of vasodilator and vasoconstrictor mediators leads to inadequate pulmonary vasodilation at birth, leading to pulmonary hypertension. Sepsis, specifically group B streptococcal infection, is associated with aPH.15 Sepsis and pneumonia result in acidosis, hypoxia, and release of pulmonary vasoconstrictor mediators such as thromboxane.16 In addition, systemic hypotension and shock secondary to sepsis decreases systemic vascular resistance (SVR), leading to right-to-left shunt and hypoxemia. Perinatal asphyxia, meconium aspiration, hypoxic-ischemic encephalopathy, and therapeutic hypothermia are commonly associated with aPH17–19 and are discussed in Chapter 22. Current strategies in the treatment of aPH focus solely on pulmonary vasodilation; effective therapies to reverse pulmonary vascular remodeling and restore angiogenesis do not receive adequate focus (Figures 25.2 and 25.3).20 In addition to pulmonary vasoconstriction, vascular remodeling and vascular hypoplasia are important contributors to the pathophysiology of aPH. Conditions such as CDH, MAS with intrauterine onset, BPD with aPH, and idiopathic (black lung) aPH demonstrate reduced blood vessel density with remodeling with hypertrophy and hyperplasia of tunica media and adventitial thickening contributing to increased PVR. This vascular remodeling is promoted by factors such as hypoxia-inducible factor-1α (HIF-1α), endothelin-1, reactive oxygen species (ROS), and PPAR-γ. In contrast, therapies that increase cAMP and cGMP in the pulmonary arterial smooth muscle cells tend to be anti-proliferative (Figure 25.2). HIF-1α is a master transcriptional regulator of developmental and cellular response to hypoxia. Among various factors, HIF-1α upregulates transcription of vascular endothelial growth factor (VEGF) and erythropoietin. Elevated HIF-1α protein has been reported in experimental models of aPH, including pulmonary arterial endothelial cells derived from lambs, with aPH induced by antenatal ductal ligation.21 The expression of hexokinase-II, a target gene of HIF-1α, is also elevated in aPH pulmonary arterial endothelial cells. Increased ROS production and oxidative stress contributes to pathophysiology of aPH and vascular remodeling.22–26 Increased ROS can inhibit prolyl hydroxylases that target HIF-1α for degradation.27–29 Reduced VEGF leads to decreased angiogenesis and vascular hypoplasia and also leads to reduced nitric oxide (NO). Reduced NO levels lead to decreased cGMP and protein kinase-G levels and are associated with decreased mitochondrial transcription factor levels, electron transport chain protein levels, vascular tube formation, and cell migration.20,30 These changes were reversed by cinaciguat (a soluble guanyl cyclase activator), 8-bromo-cGMP, and sildenafil (a phosphodiesterase-5 inhibitor).20 Reduced AMP kinase (AMPK) activity contributes to impaired mitochondrial function, reduced eNOS, and decreased branching and vessel formation in lambs with aPH induced by antenatal ductal ligation.31 In utero treatment of these lambs with metformin stimulates AMPK and restores vessel density in the lungs of these lambs.31 Rho kinase (ROCK) signaling is a complex pathway responsible for smooth muscle contraction, cellular proliferation, migration, differentiation, and gene expression in various vascular beds, including the neonatal pulmonary circulation.32–34 In experimental models of aPH increased ROCK activity increases calcium sensitization and vascular tone and contributes to hypertensive remodeling mediated through endothelin-1 and peroxisome proliferator-activated receptor-γ (PPAR-γ) pathway.32,33 PPAR-γ is a member of the nuclear receptor superfamily, decreases pulmonary arterial smooth muscle cell (PASMC) proliferation, and is abundantly expressed in vascular smooth muscle cells and endothelial cells in the lung.35 Agonists of PPAR-γ decrease ROCK activity and reduce proliferation of PASMC.32 The cellular mechanisms responsible for vascular hypoplasia and remodeling are shown in Figure 25.3. Agents that increase cGMP (sildenafil, cinaciguat and NO),36,37 those that decrease ROS (antioxidants),25,38 ROCK inhibitors (Y-27632, fasudil),34 AMPK stimulators (metformin),31 endothelin receptor blockers (bosentan),33 and PPAR-γ agonists (rosiglitazone)32 have the potential to promote pulmonary vasodilation and reverse vascular modeling in aPH and are being currently used or investigated as potential therapeutic agents. Acute hypoxemic respiratory failure with persistent hypoxemia is a hallmark feature of aPH and differentiating cyanotic CHD from acute PH is critical in a hypoxemic cyanosed infant. The initial evaluation should include a thorough history of risk factors for aPH, a meticulous physical examination, simultaneous measurement of pre-ductal (right upper limb) and post-ductal (lower limb) oxygen saturation to check the difference between them, chest radiography, and arterial blood gas analysis. Pre- and post-ductal oxygen saturation and PaO2 measurements can help in differentiating aPH from cyanotic CHD. Pre-/post-ductal saturation differences of greater than 5–10% or PaO2 differences of 10–20 mmHg between right upper limb and lower limbs, with pre-ductal levels being higher than post-ductal levels, are considered significant. The lack of a pre-/post-ductal saturation difference does not exclude a diagnosis of aPH, as the ductus arteriosus must be patent for this feature to be noted. Hypoxemia is often labile and variable in aPH, unlike the fixed hypoxemia seen in cyanotic CHD.4,39 The chest X-ray is particularly helpful in diagnosing respiratory pathology. It may help in differentiating the etiology of aPH (such as MAS, pneumonia, RDS, CDH, etc.) and may also help in differentiating types of CHD.40,41 Hypoxemia disproportionate to the severity of parenchymal disease on chest radiography suggests idiopathic PH or underlying cyanotic heart disease (Figure 25.1). Pulmonary oligemia is seen in tetralogy of Fallot (TOF), Ebstein anomaly, critical pulmonary stenosis, and pulmonary atresia due to decreased pulmonary flow. Pulmonary plethora is seen in transposition of great arteries (TGA) with intact interventricular septum, truncus arteriosus, tricuspid atresia, total anomalous pulmonary venous connection (TAPVC), and single ventricle.41 From a pulmonary management perspective, aPH secondary to parenchymal lung disease needs optimal PEEP or mean airway pressure to expand the lung to FRC and to prevent expiratory airflow obstruction in TTN and MAS. In contrast, aPH secondary to pulmonary hypoplasia associated with CDH or oligohydramnios may benefit from lung protective ventilation (with relatively low PEEP).42 Finally, infants with idiopathic aPH may benefit more from early use of pulmonary vasodilators.4 The hyperoxia test may be useful in differentiating the cardiac causes from respiratory causes in cyanotic newborns. On confirmation of central cyanosis by measuring the arterial partial pressure of oxygen (PaO2), response of PaO2 to 100% oxygen inhalation is tested (hyperoxia test). Oxygen should be administered through a plastic hood for at least 10 minutes in order to fill the alveolar spaces completely with oxygen. In a cyanotic CHD the rise in PaO2 is usually no more than 10–30 mmHg and hardly ever exceeds 100 mmHg. With pulmonary diseases, PaO2 often rises greater than 100 mmHg. However, infants with massive intra-pulmonary shunt from severe respiratory disease may not show a rise in PaO2 to 100 mmHg. Conversely, some infants with cyanotic defects with a large pulmonary blood flow, such as TAPVC, may demonstrate a rise in PaO2 of 100 mmHg or higher. A hyperoxia test should be interpreted in the context of the clinical picture and the degree of pulmonary pathology seen on x-ray.41,43 With the availability of bedside echocardiography, the hyperoxia test is rarely performed but it is still a very helpful bedside test when echocardiography is not available. Echocardiography remains the gold-standard bedside test to confirm the diagnosis of acute PH, monitor the response to the therapeutic interventions, and rule out underlying cyanotic or critical CHD. It can help in confirming the diagnosis of acute pulmonary hypertension and to assess its severity. In the absence of right ventricular (RV) outflow obstruction, pulmonary artery systolic pressure (PASP) can be estimated using tricuspid regurgitation velocity or ductus arteriosus shunt when present. The pulmonary artery systolic pressure (PASP) can be estimated using echocardiography in the presence of tricuspid regurgitation (TR), as demonstrated (Figure 25.4). The pressure gradient between the RV and right atrium (RA) is calculated by using the modified Bernoulli’s principle.44,45 PASP is estimated by adding RA pressure, which is usually about 5 mmHg, to the peak pressure gradient between RV and RA obtained on continuous wave (CW) Doppler, when there is no RV outflow tract obstruction (Figures 25.4 and 25.5). However, pulmonary pressures may be underestimated if there is impaired RV contractility, and TR may be absent in up to one-third of infants with aPH.
Chapter 25: Pathophysiology and assessment of acute pulmonary hypertension of the newborn
Introduction
Classification of aPH
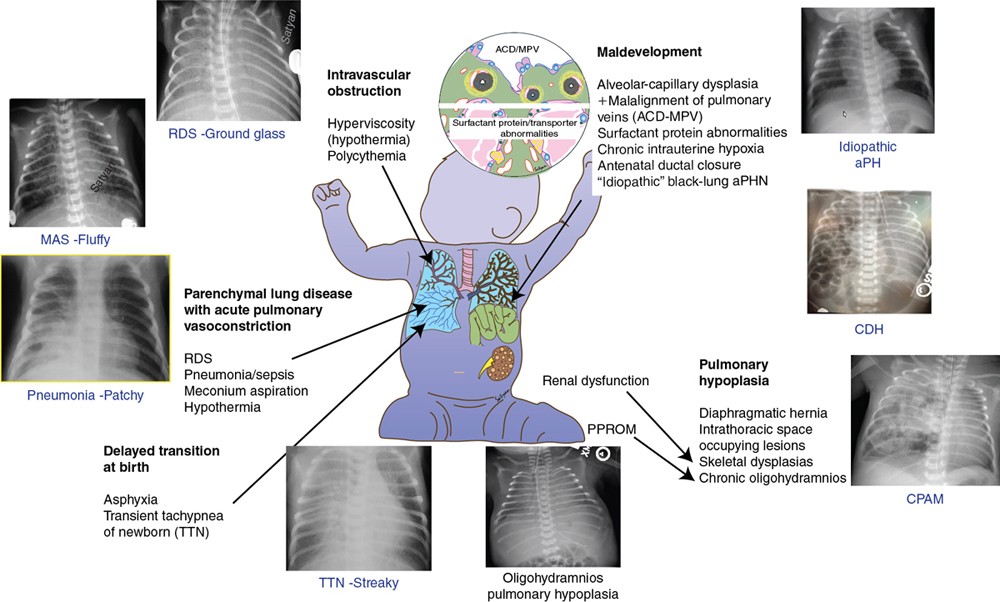
Pathophysiology of acute pulmonary hypertension in the newborn
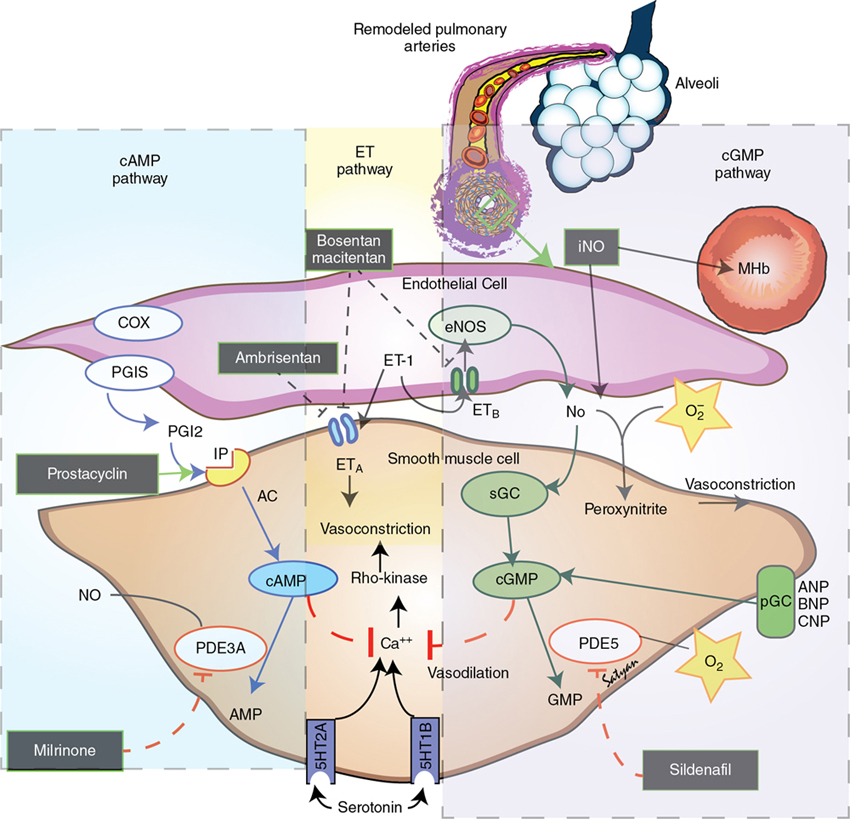
Mechanisms of pulmonary vascular remodeling
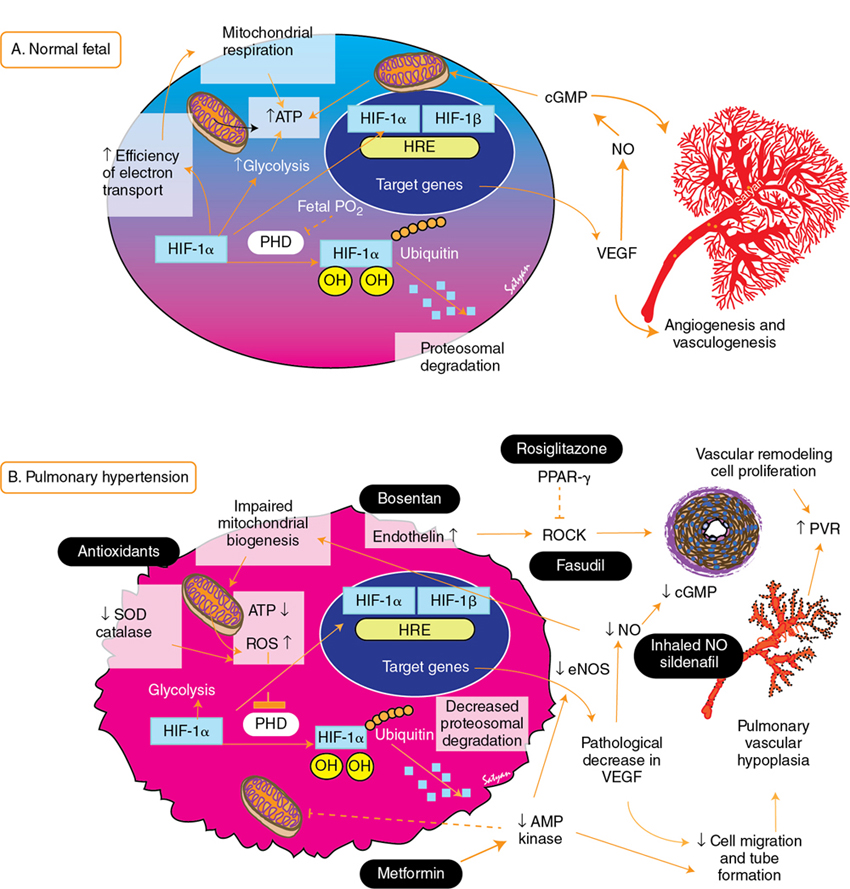
Clinical presentation and diagnosis of acute pulmonary hypertension in the newborn
Echocardiographic evaluation of acute pulmonary hypertension of newborn

![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathophysiology and assessment of acute pulmonary hypertension of the newborn
