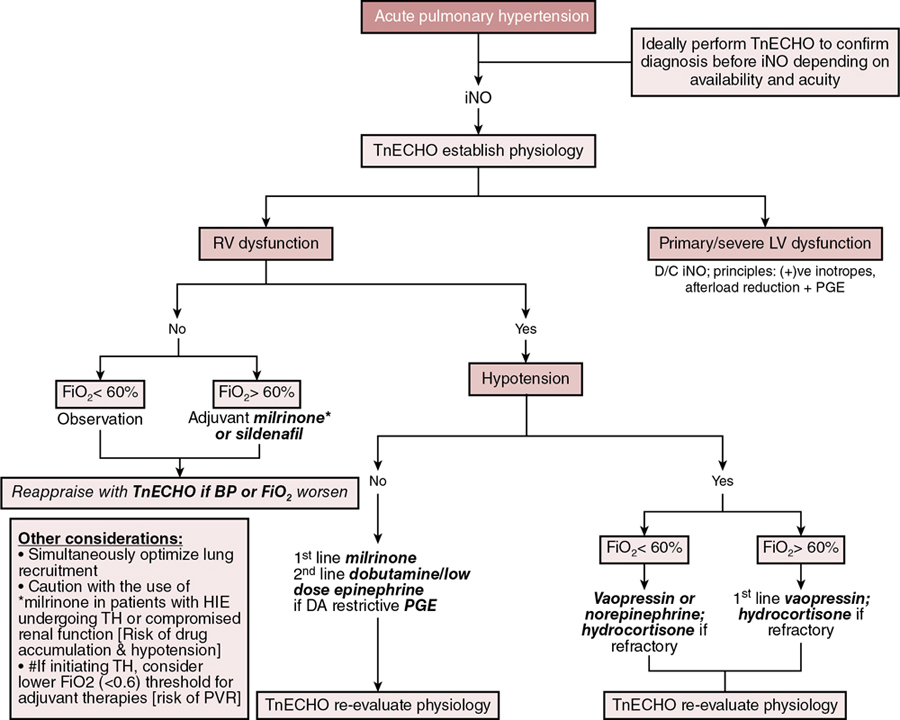
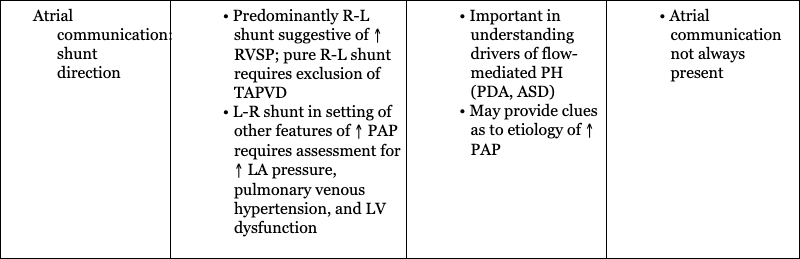
Stephanie M. Boyd, Trassanee Chatmethakul, Patrick J. McNamara Key points Neonatal pulmonary hypertension is a heterogenous condition affecting both term and preterm neonates with a range of underlying disease processes, the hallmark of which is elevated pulmonary arterial pressure (PAP). Mean PAP (mPAP) is dependent upon pulmonary vascular resistance (PVR), cardiac output (CO) (and shunts), and left atrial pressure. Calculation of mPAP is possible, using pulmonary capillary wedge pressure (PCWP) as a proxy for left atrial pressure: mPAP = [PVR × CO] + PCWP The primary pathophysiologic event in aPH occurring during the perinatal transition period, classically referred to as persistent pulmonary hypertension of the newborn (PPHN), is interruption to or failure of the normal postnatal decline in pulmonary vascular resistance (PVR). This may be due to maladaptation (e.g., perinatal asphyxia, meconium aspiration syndrome), maldevelopment of the pulmonary vasculature (e.g., in utero closure of the ductus arteriosus), or pulmonary underdevelopment or hypoplasia in the setting of e.g., congenital diaphragmatic hernia or oligohydramnios. Whatever the underlying etiology, there is associated dysregulation of the pulmonary vascular bed and changes in cardiac loading conditions, with significant hemodynamic consequences. Although PPHN is a commonly used descriptor, aPH may be a more appropriate term for the constellation of features that include hypoxemia and right ventricular dysfunction, due to the protracted time taken for physiological decline in PVR after birth in healthy newborn infants. Persistent elevations in PAP after birth are essentially ubiquitous, and for healthy infants, not pathological. In the setting of aPH, the resultant low oxygen saturation is poorly tolerated after birth and affected neonates may develop multi-organ dysfunction/failure because of hypoxemia and inadequate tissue perfusion. The hemodynamic consequences of aPH (Chapter 25) may include right ventricular dysfunction, dilation and hypertrophy due to increased afterload, and impairments in pulmonary blood flow. Adverse systemic effects include hypoxemia, abnormal left ventricular (LV) mechanics, and low cardiac output. Although generally considered to be a primary disorder of elevated PVR, it is evident from the “mPAP equation” that elevations in cardiac output and/or left atrial pressure may also result in raised pulmonary pressure, with implications for management. Accurate delineation of the pathology requires comprehensive hemodynamic assessment and a detailed understanding of the interplay of the neonatal cardiovascular system with the various underlying disease processes (Table 27.1). Adapted from Giesinger R, Kinsella JP, Abman SH, McNamara PJ. Pulmonary hypertension phenotypes in the newborn. In: Dakshinamurti S, ed. Hypoxic respiratory failure in the newborn: from origins to clinical management. Boca Raton: Taylor & Francis Group; 2021. The clinical manifestations of aPH depend on the degree of elevation of PAP; the presence, direction, and magnitude of shunting through the patent ductus arteriosus (PDA) and/or patent foramen ovale (PFO); right and left ventricular performance and ability to adapt to changes in cardiac loading conditions; and the presence of associated morbidities. The severity of aPH can run the full clinical spectrum, from mild hypoxemia with varying degrees of respiratory distress to severe hypoxemia and cardiopulmonary instability necessitating advanced intensive care support. Hypoxemia in the newborn may result from lung parenchymal and/or cardiovascular pathology, and differentiating the contributors to poor oxygenation clinically can be challenging. Multiple lung and vascular pathologies may also coexist in the same patient. For example, in the setting of aPH secondary to meconium aspiration, atelectasis, intrapulmonary shunting, alveolar edema, and hyperinflation may all be present together with pulmonary vascular remodeling, hypoxic vasoconstriction, and myocardial dysfunction due to perinatal hypoxia-ischemia. Hypoxemia out of proportion to the degree of underlying lung disease and lability in oxygenation are both characteristic of aPH, and presence of either of these clinical features should raise suspicion of a cardiopulmonary vascular component to the infant’s presentation. Approaches to treatment vary according to the severity of the pathophysiology, the underlying cause(s), and the hemodynamic phenotype. Consequently, an in-depth understanding of the pathophysiology (Figure 27.1) is essential to allow individualization of therapy and appropriate targeting of treatment modalities. Due to the complexity of interactions between PVR, lung aeration, and myocardial adaptation, perinatal transition is a time of heightened vulnerability for the newborn cardiovascular system. Perturbations in this physiological framework due to any combination of perinatal hypoxia-ischemia, infection, and/or pulmonary parenchymal disease may interfere with the normal physiological decline in PVR and result in development of aPH. As a result of the elevated PVR in arterial (classic) aPH, hypoxic vasoconstriction ensues, which is an adaptive response to optimize ventilation-perfusion (V/Q) matching.1 Poor lung recruitment and overdistension from positive pressure ventilation can worsen hypoxic vasoconstriction, due to both increases in PVR and impaired carbon dioxide clearance, resulting in acidosis. It is important to realize that PVR is lowest at optimal lung recruitment, that is, at or near FRC.2 With excessive lung volume, some pulmonary capillaries undergo narrowing and stretching, whereas reduced lung volume can lead to capillary tortuosity or kinking, both of which increase PVR.2 Careful titration of positive pressure ventilation based on clinical, blood gas, and radiological assessment is therefore necessary. Particular care should be taken to avoid lung overinflation in the setting of hypoxic-ischemic encephalopathy, where lung compliance and FRC may be normal and hypoxemia instead a manifestation of altered pulmonary vascular reactivity. Overly aggressive attempts at lung recruitment in this context may be complicated by further impairments in PBF,3 as well as systemic compromise from reductions in LV stroke volume and LV end-diastolic volume (LVEDV).4 Over-distension also has the potential to compromise pulmonary venous flow, which may be difficult to distinguish clinically from progression of aPH driven by high PVR.5 Additional non-pulmonary factors related to either the underlying disease process, such as sepsis, perinatal asphyxia, and hypovolemia, or iatrogenesis, such as treatment with vasopressors that can cause pulmonary vasoconstriction (e.g., high-dose dopamine), may further drive increases in PVR. Elevated PVR leads to increased RV afterload6 and bidirectional or right-to-left shunting through the PFO and PDA. Right-to-left atrial shunting contributes to a reduction in RV preload.7 Right-to-left ductal shunting, though contributing to hypoxemia and reduced PBF, may provide a pop-off mechanism for the RV exposed to high afterload and an additional source of post-ductal perfusion where there is significant LV impairment. Elevated pulmonary pressure results in a reduction in the pressure gradient between the RV cavity and the pulmonary vascular bed. The initial increase in RV afterload results in compensatory RV hypertrophy and increased contractility (referred to as “coupling” or homeometric adaptation).8 Increased contractility is mediated by changes in the sarcomere length-tension relationship and cardiomyocyte force-frequency relationship and increased calcium sensitivity.9 The force-frequency relationship describes the adaptive response of the neonatal myocardium to tachycardia, leading to improved force generation with increasing heart rate.10 To maintain cardiac output in the face of increased afterload, the RV dilates and heart rate increases. These changes preserve stroke volume initially by way of the increase in stroke volume associated with increased preload – via the Frank-Starling mechanism, with a larger ventricle emptying proportionally less blood – and an intact force-frequency response. This process is referred to as heterotopic adaptation.11 Unable to generate sufficient pressure for complete ejection of blood with each cardiac beat, RV emptying becomes progressively inadequate, leading to an increase in RV end-systolic pressure and, because less blood is ejected during systole, an increase in RV end-diastolic pressure.11 In addition, the force-frequency response is blunted as cardiac dysfunction develops. Progressive dilatation increases wall stress and myocardial oxygen consumption and eventually leads to a reduction in stroke volume.8 RV dilatation also changes the configuration of the cardiac chamber into one that is more spherical, which is associated with functional regurgitation across the tricuspid valve and increased right atrial pressure.12 Where elevation of PVR is excessive and sustained, ischemia, hypoxemia, and myocardial stretching lead to progressive RV dysfunction and uncoupling.8,13 This is associated with leftward bowing of the interventricular septum (IVS), which is caused by prolonged contraction of the RV free wall when compared with the septum or LV free wall, impairing diastolic LV filling.14 Together with RV dilation, this leads to further impairment of RV function and decreased PBF.15 Through ventricular interdependence, the leftward shift in septal configuration also reduces LVEDV, LV stroke volume,9 and RV systolic contractile force,16,17 resulting in systemic hypoperfusion, acidosis, and further reductions in PBF. In some infants a cycle of reduced PBF, worsening acidosis, hypoxemia, ventilation-perfusion mismatch, and cardiac dysfunction may develop, with a clinical phenotype of hypoxemic respiratory failure and cardiocirculatory impairment. Increased pulmonary blood flow, such as that observed in left-to-right shunt lesions, may result in elevations in pulmonary arterial pressure. The most obvious mechanism for this is an increase in pressure driven by increased flow through a circuit with a finite capacity to expand to accommodate the additional circulating volume. Muscular hypertrophy of the pulmonary vasculature, with secondary elevation in PVR, also occurs in response to a sustained increase in PBF.18,19 An additional contributor is a disturbance of endothelial function resulting in altered pulmonary vasoreactivity; specifically, selective impairment of endothelium-dependent pulmonary vasodilation. Based on animal models, this may be attributable to both high basal activity of nitric oxide (NO) that is not readily increased20 and dysregulation of endothelial NO synthase,21 induced by increased pulmonary blood flow and pulmonary hypertension. This attenuates the fetal endothelial release of NO in response to sheer stress22 such as that generated by excessive pulmonary blood flow and may contribute to elevated mPAP in this setting. Newborns with large preductal systemic to venous connections resulting in high-volume shunts are particularly at risk of complications from right heart pressure and volume loading. For example, the vein of Galen aneurysmal malformation (VGAM) is a rare cerebral arteriovenous malformation (AVM) with major hemodynamic implications. In utero, the low resistance of a cerebral AVM is balanced by the low-resistance placental circuit. With removal of the placenta and transition to the high afterload systemic neonatal circulation, up to 70% of the cardiac output is directed toward the cerebral vasculature.23 Superior vena caval flow may be up to 10 times normal, reflecting high flow through the AVM.24 This may result in a profound reduction in lower body perfusion, with lactic acidosis and post-ductal arterial hypotension. The ventricles receive increased preload, which in the case of the RV must be ejected against increased afterload, predisposing to RV dysfunction and failure. Supra-systemic pulmonary hypertension occurs,24 which is associated with right to left shunting at atrial and ductal levels.23,24 This perpetuates hypoxemia, which further compromises ventricular performance. Chronic volume and pressure loading leads to RV dilation, which increases susceptibility to afterload-mediated dysfunction25 as adaptive mechanisms fail.17 RV coronary perfusion normally occurs throughout systole and diastole and is characterized by a lower flow and oxygen extraction when compared with the LV.17 The maladaptive, dilated, and hypertrophied RV has a higher oxygen requirement and inefficient oxygen utilization,17 with coronary perfusion limited to diastole by the increased RV pressure.26 Elevations in RV end-diastolic pressure, leading to increased right atrial and central venous pressure, and therefore reduced diastolic coronary flow to the RV constitute a potential additional source of impaired perfusion in the setting of PH.11 Intracranial vascular “steal” through the AVM, which is associated with retrograde diastolic descending aortic flow, may further compromise tenuous coronary perfusion via low coronary root pressure, creating a “pseudocoarctation” physiology.27 This effect is additive with the increased ventricular pressure in reducing subendocardial perfusion, contributing to myocardial ischemia and RV dysfunction.23 Non-judicious use of pulmonary vasodilators may exacerbate these effects, particularly in the setting of an open ductus. Despite the increase in PVR, the shunt in VGAM is obligatory, such that pulmonary vasoconstriction does not offset the high-output cardiac failure, as might occur in congenital cardiac disease. This is postulated as a contributor to the severity of cardiac failure in infants with VGAM.28 Poor function of the left ventricle results in inefficient ejection of end-diastolic volume, which results in a progressive increase in left ventricular end-diastolic pressure (LVEDP). This leads to pulmonary venous congestion, increased pulmonary capillary pressure, and a hydrostatic gradient across the interstitium, resulting in fluid transudation to alveolar spaces, i.e., pulmonary edema. Similar to left-to-right shunt lesions, endothelial dysfunction is also implicated as contributory to elevated pulmonary pressure in obstructive pulmonary venous hypertension.29 Left and right ventricular dysfunction may coexist in transitional aPH, although there are subgroups in whom LV disease may occur independently. Fetal and neonatal echocardiographic studies of infants with congenital diaphragmatic hernia (CDH) support disturbances of fetal LV development and postnatal LV dysfunction as part of the pathogenesis of hypoxemia in affected infants.30–33 Early LV systolic function correlates with markers of clinical disease severity in CDH and may be a primary determinant of illness severity rather than a secondary consequence of cardiovascular instability.30 These findings have implications for treatment, and use of pulmonary vasodilators, since increasing pulmonary blood flow and LV preload may risk exacerbating existing LV diastolic dysfunction and pulmonary venous hypertension.34,35 Flash pulmonary edema has been observed with pulmonary vasodilator therapy in pulmonary venous disease.36 Conversely, improvements in RV performance by afterload reduction may indirectly augment LV performance through ventricular interdependence.30 A multicenter randomized trial of sildenafil and inhaled NO in CDH (CoDiNOS trial) utilizing cardiac function assessment may provide additional guidance on use of pulmonary vasodilators in this setting.37 In general, pulmonary vasodilators should be used with caution in infants with LV dysfunction and are likely to be harmful where LV impairment is severe. The pathophysiology of cPH (Chapter 26) is influenced by the development of the pulmonary vasculature and the status of the lung parenchyma.15 Maldeveloped pulmonary vasculature with abnormal lung parenchyma and elevated PVR is typical of BPD and pulmonary hypoplasia. Maldeveloped pulmonary vasculature is less commonly encountered and is observed in, for example, alveolar capillary dysplasia (ACD) and primary surfactant deficiencies. ACD with misaligned pulmonary veins may, however, account for a greater proportion of unexplained PH in infants than previously thought.38 Chronic PH secondary to pulmonary venous congestion may be due to either increased PBF (e.g., left-to-right shunts), or increased pulmonary capillary wedge pressure).15 Elevated PCWP, or left atrial pressure, in the newborn most commonly occurs due to LV dysfunction or acquired pulmonary vein stenosis, although left-sided cardiac lesions may also produce this cPH phenotype. Chronic PH associated with BPD (BPD-cPH) is characterized by abnormal remodeling and growth arrest of the pulmonary vasculature,39,40 and impaired angiogenesis and alveolarization,39,41 resulting in abnormal pulmonary vascular function with increased PVR, high RV afterload, and RV dysfunction. Abnormal pulmonary vascular remodeling is triggered by postnatal hypoxia/hyperoxia.39 Additive insults, such as endothelial dysfunction, and inflammatory stimuli, such as infection and ventilator-induced lung injury, are thought to perpetuate alveolar hypoxia39,40,42 in at-risk infants. Antenatal factors, including inflammation, intrauterine growth restriction, and the lung microbiome,43 are also implicated in the pathogenesis. There is evidence that echocardiographic evidence of PH within the first 7–14 days after birth may predict subsequent moderate-severe BPD or death at 36 weeks postmenstrual age,44,45 underscoring the importance of abnormal pulmonary vascular development in the development of BPD. Conversely, a very low risk of late PH has been observed in the absence of PH at 7 days of age.45 The approach to management includes lung optimization alongside treatment with pulmonary vasodilators (e.g., sildenafil). Both clinical improvement and reductions in pulmonary artery pressure have been documented with sildenafil therapy.46 In the presence of RV dilatation, diuretics may also be considered for optimizing RV configuration.5 Although long-term benefits of diuretics have not been demonstrated in infants with BPD, improvements in both oxygenation and pulmonary mechanics have been observed.47 In addition, a retrospective study demonstrated high rates of symptomatic improvement in BPD-cPH, with diuretic treatment where RV dilatation was used as the treatment threshold.48 Sustained left to right shunt and resultant pulmonary over-circulation may occur in particular at atrial level (e.g., PFO or atrial septal defect [ASD]) or at ductal level,5 where a PDA persists over an extended period of time. Chronic over-circulation results in pulmonary vascular remodeling, which is also a potential driver of eventual PDA closure.49 Elevations in pulmonary arterial pressure are initially due to increased PBF and, with chronicity, result in changes to pulmonary vascular architecture and reactivity.50,51 Both shear stress injury and endothelial dysfunction are implicated in the pathogenesis,52 as in acute flow-driven PH. In high-volume shunts leading to unrestrictive pulmonary artery flow, increases in PVR may eventually be sufficient to lead to PDA shunt reversal in some patients.50,51 Infants with coexistent ASD and PDA, or other multi-level shunts, are at greatest risk of abnormal pulmonary physiology due to dual sources of over-circulation.5 This is highlighted by the 2.44-fold increase in PH among preterm infants with an ASD in an echocardiographic study of 334 infants born at <32 weeks’ gestation.53 Pulmonary vasodilator use in this setting may be harmful both due to the drop in PVR resulting in an increased SVR:PVR ratio and the risk of contributing to oxidative stress.5 Adequate shunt management in the mainstay of therapy. In the case of PDA this may include pharmacological or interventional (surgical or transcatheter) closure. Although diuretics are often used for symptomatic management, historical studies of furosemide have noted prostaglandin-mediated dilatation of ductus arteriosus vessels in an animal model54 and an increased incidence of PDA.55 While judicious fluid management is sensible, aggressive fluid restriction has been associated with a greater risk of acute kidney injury56 as well as concerns regarding nutritional intake. In addition, reducing overall circulating fluid volume without any effect on the proportion of cardiac output directed toward the lungs poses a risk of exacerbating existing post-ductal hypoperfusion from PDA run-off.57 Conversely, diuretics are a key aspect of management for patients with ASD, with device closure performed successfully in infants as small as 2–2.5 kg.58 While cPH and right ventricular function are the usual focus of cardiovascular sequelae of BPD, there is increasing interest in the contribution of LV disease to BPD-related symptomatology.59–62 Clinical identification of preterm born infants with respiratory morbidity and comorbid LV pathology is challenging, since features of post-capillary pulmonary venous hypertension closely mimic those of BPD with primary parenchymal pathology. Persistence of pulmonary edema despite diuresis and worsening of pulmonary edema in response to pulmonary vasodilator therapy are “red flags” for underlying LV disease.61 In association with LV mal-compliance, high rates of systemic hypertension have been observed among infants with BPD.63–65 The increased afterload imposed by aortic stiffness associated with systemic hypertension (loss of Windkessel effect) is postulated to produce sufficient back pressure over time to result in disturbances of left heart function and pulmonary venous hypertension.61 Additionally, polymorphisms in angiotensin-converting (AT-converting) enzyme genes have been identified in infants with CDH and features of PH66 and alterations in the AT-II receptor pathway have been shown to reduce hyperoxia-mediated heart and lung injury in a neonatal rat model.67 The role of AT-II in modulating blood pressure via both increasing SVR and stimulating release of norepinephrine and vasopressin highlights the potential for a neurohormonal contribution to neonatal hypertension and BPD.5 Furthermore, the effectiveness of an endothelin receptor antagonist in reducing PH and RV hypertrophy in a neonatal rat model provides additional translational data as to the importance of the endothelium in the pathogenesis of PH in newborns.68 There is preliminary evidence that improvement of cardiorespiratory indices, and diastolic function in particular, may be achieved by treatment with ACE inhibitors.69 RV output has been shown to be increased, accompanied by a significant lowering of PVR with ACE inhibition in a small case series of infants with severe BPD unresponsive to conventional therapies.69 Additionally, improvement in LV diastolic function indices with ACE inhibitor treatment has been observed in neonates with systemic hypertension and LV diastolic dysfunction.70 Anti-hypertensive therapy with ACE inhibitors is suggested as a treatment option for LV disease in this patient group, with appropriate monitoring of renal function and caution with co-administration of diuretics.5 Due to the risk of precipitation of pulmonary edema and worsening of pulmonary venous hypertension, pulmonary vasodilator therapy is best avoided in preterm born infants with chronic PH of this nature, although in some patients true pulmonary arterial and venous hypertension may coexist, requiring combination therapy. Assessment of underlying pathophysiology in PH may be challenging clinically as labile hypoxemia and comorbid pulmonary disease are common to several hemodynamic phenotypes. The final common pathway is high RV afterload leading to RV dysfunction, with or without systemic effects from disturbances in LV morphology and function. Delineating flow-driven PH from that caused by primary elevations in PVR or mal-compliance of the LV is important for instituting disease-specific management. For example, pulmonary vasodilator therapy may be harmful in the context of pulmonary venous hypertension from LV disease. Targeted neonatal echocardiography (TnECHO) may aid in adjudication of the underlying cause(s) of PH, as well as titrating treatment against clinical response.71,72 The approach to a newborn with suspected PH includes confirmation of the elevation in mPAP; assessment of PVR and right ventricular performance; exclusion of major structural congenital cardiac disease that can mimic primary PH; and evaluation of LV function, systemic blood flow, and the magnitude and directionality of intracardiac shunts. Qualitative measures of RV size and function, even by trained evaluators, are unreliable,73 and biventricular cardiac dysfunction is common.74 Comprehensive appraisal of both RV and LV performance is recommended. A number of congenital cardiac defects, including total anomalous pulmonary venous drainage (TAPVD) and transposition of the great arteries (TGA), may present with severe hypoxemia and mimic aPH. Presence of labile hypoxemia and/or systemic hypotension makes aPH more likely75; however, this is not absolute and distinguishing between aPH and congenital cardiac lesions on clinical grounds can be challenging. Initiation of prostaglandin E1 while awaiting definitive anatomical assessment may be appropriate depending on the clinical presentation. Specialist advice should be sought early, and cardiac structural evaluation should occur as part of the first echocardiogram, or at the earliest opportunity.76 This is important so as not to delay specialist cardiology input and care in a cardiac surgical center, as well as because some treatments for aPH, such as pulmonary vasodilator therapy, can worsen hemodynamics in subgroups of infants with congenital cardiac disease. Typical findings in resistance-mediated aPH include RV systolic and diastolic failure due to increased afterload, decreased RV stroke volume and filling, decreased PBF with V/Q mismatch, RV dilatation resulting in a D-shaped left ventricle with a decrease in LV preload and stroke volume, and right-to-left ductal shunting through the PDA and/or PFO.76 TnECHO, also referred to as clinician-performed ultrasound, is a useful adjunct to clinical assessment. It may be utilized to confirm the diagnosis of aPH and disease severity, delineate the underlying pathophysiology to guide treatment selection and therapeutic target(s), monitor response to therapy, and wean supportive treatment as the infant’s condition improves. Echocardiographic measurements commonly employed in assessment of aPH are outlined in Table 27.2. RVSP using TR jet (mmHg) Direct measure of RV systolic pressure Moderate correlation with invasive measures of RVSP77 Septal curvature – subjective assessment Assessment of RV versus LV pressure Easily measured and universally present Eccentricity index Assessment of RV versus LV pressure Reproducible, validated in newborns PAAT PAAT:RVET (ms) Mid-systolic notching of PA Doppler waveform Serial assessment to monitor PVR over time or with treatment Easily measured RVO (mL/kg/min) Marker of pulmonary blood flow and RV function TAPSE (mm) Measure of RV function RV FAC Measure of global RV function Normative data exist for newborns79 TDI of IVS and RV free wall Assessment of systolic and diastolic function May be useful for monitoring response to treatment LVO (mL/kg/min) Marker of systemic blood flow and LV function PDA: ratio of pulmonary artery to aortic pressure (PAP:AOP) based on shunt direction: Uses Doppler of ductus arteriosus Direct comparison of pulmonary to systemic arterial pressure Useful for qualifying RVSP as subsystemic, systemic, or supra-systemic Atrial communication: shunt direction Adapted from Jain A, Giesinger RE, Dakshinamurti S, et al. Care of the critically ill neonate with hypoxemic respiratory failure and acute pulmonary hypertension: framework for practice based on consensus opinion of neonatal hemodynamics working group. J Perinatol. 2022;42:3–13. Pulmonary artery pressure may be estimated by several different methods. These are discussed in detail in Chapters 9 and 10 and include tricuspid valve regurgitation (TR) peak velocity, PDA right-to-left flow peak velocity, interventricular septum (IVS) configuration, and LV systolic eccentricity index (LV-sEI).76 For estimation of PAP using the TR jet, the angle of insonation should be less than 20° to avoid underestimation of systolic pulmonary artery pressure. The modified Bernoulli equation is used and right atrial pressure, though not measured, is assumed to be 3–5 mmHg.76 Assessment of the TR jet in multiple views is advantageous in this regard. The measurement is generally reliable if accurate measurement is taken using a full Doppler spectral envelope, with moderate correlation observed with catheter laboratory measures of PAP.77 Tricuspid regurgitation is present in approximately 70% of infants with aPH.80 Absence of a measurable TR velocity does not imply absence of PH,81 and evaluation of PAP using this method is not reliable in the presence of right ventricular outflow tract obstruction or RV failure.76 Pulmonary regurgitation peak velocity can also be used to estimate PAP using the modified Bernoulli equation, with RV diastolic pressure assumed to be 2–5 mmHg. Although PAP may be estimated using transductal right-to-left flow peak velocity if right-to-left shunting occurs for ≥30% of the cardiac cycle, this is often not reliable,76 and assessment of direction of the ductal shunt is probably more useful. Configuration of the IVS, either qualitatively or using the LV-sEI, can also be used to estimate PAP. Normal ventricular geometry consists of an O-shaped LV, with an RV systolic pressure (RVSP) of <50% of LV pressure (LVP). With increased pulmonary pressure, flattening of the IVS occurs, resulting in a loss of circularity and D-shaped LV (estimated RVSP of 50–100% of LVP). With further increases in RV afterload, the RV eventually curves into the LV, producing a crescent-shaped LV and an estimated RVSP that is systemic or supra-systemic (≥100% of LVP).76,82 Changes in IVS configuration relative to the cardiac cycle can provide some insight as to the likely underlying pathophysiology. In resistance-driven aPH due to elevated PVR, bowing of the IVS occurs due to prolonged contraction of the RV free wall (against the increased afterload) when compared with the septum or LV free wall.14 This results in predominant flattening or bowing of the IVS during systole and impairment of LV diastolic filling.14 Conversely, in the setting of flow-driven PH secondary to a left-to-right shunt lesion, the RV pressure relative to the LV is higher in diastole, due to increased filling from left-to-right shunt. The LV-sEI provides an objective estimate of IVS flattening/bowing76 and uses the end-systolic ratio of the anterior-inferior and septal-posterolateral LV cavity dimensions, respectively. Interobserver agreement is higher for LV-sEI than for subjective assessment of septal configuration83 and values of ≥1.3 show strong correlation with catheterization laboratory measurements with good sensitivity and specificity.84 The normal value is 1, and more than half-systemic RV pressure has been demonstrated at values of ≥1.3 in an echocardiographic study of 216 newborns at risk of PH.83 Right ventricular systolic time intervals have been validated for estimation of PVR,85 including in newborns.86 A pulmonary artery acceleration time (PAAT), also referred to as time to peak velocity (TPV), of <90 ms identifies PH with a sensitivity of 97% and specificity of 95%.85 A PAAT to RV ejection time (RVET) ratio of <0.31 also reliably detects PH, with a value of <0.23 suggestive of marked elevation of PAP.85 Additional measures of PVR, such as the tricuspid regurgitation velocity to RV outflow time-velocity integral (TRV/VTI[RVOT]) and pulmonary artery compliance, may also be used; however, these are outside the scope of this chapter. Impaired myocardial performance results in dilatation of right heart structures, and this is visible qualitatively during echocardiographic assessment. Although accuracy is marginally greater in the hands of expert assessors, reliability of qualitative RV evaluation in newborns has not been established73 and should not be used in isolation. Appraisal of RV function is a crucial aspect of hemodynamic assessment in aPH. Reductions in several RV function measures have been associated with poor outcome in infants with aPH, including tricuspid annular plane systolic excursion (TAPSE) and RV global longitudinal peak strain.87 A TAPSE of <4 mm has been shown to be predictive of ECMO or death,87 and in a retrospective study TAPSE and LV systolic velocity were lower in patients who subsequently died or required ECMO.74 Neonatal normative values have been published for TAPSE,78 although limitations include angle and load dependency.76 In infants with comorbid HIE undergoing therapeutic hypothermia, lower RV and LV systolic and RV diastolic performance have been demonstrated,74 and cerebral function and oxygenation are correlated with RV function.88 Importantly, impaired RV performance in this population has been associated with adverse outcomes,89 including an independent association between TAPSE <6 mm and a composite outcome of death, diagnosis of cerebral palsy, or low developmental assessment scores.90 In addition, early echocardiography evidence of aPH and RV dysfunction have been associated with death and adverse outcomes, including requirement for ECMO, in infants with CDH.91 RV fractional area change (FAC) is a quantitative measure for which normative values in term and preterm newborns (25–45%) have been published.92 Values of 19% have been associated with death or a requirement for ECMO.87 Detailed RV function assessment is reviewed in Chapters 10 and 11. Newer measures, such as RV longitudinal strain using speckle tracking echocardiography (STE), show greater correlation with cardiac magnetic resonance (MR) RV ejection fraction and may be more sensitive in detection of myocardial dysfunction at earlier stages of disease93 – these techniques are discussed in detail in Chapter 11. A reduction in both RV and LV global longitudinal strain have been observed in neonatal aPH74 and RV function in response to iNO treatment has been monitored longitudinally using STE.94 In term infants with PPHN who were iNO non-responders, RV strain and strain rates improved over a 24-period following administration of milrinone,95 highlighting their utility in identifying and monitoring cardiac dysfunction in aPH. Diastolic function may be studied using tissue Doppler by measuring the tricuspid inflow velocities (Early [E], Late [A] and ratio [E:A]) during diastole, performed from the apical four-chamber view using PW Doppler. Peak systolic (s′), early diastolic (e′), and late diastolic (a′) myocardial velocities can be measured using TDI, as well as the systolic to diastolic (S/D) ratio and isovolumic relaxation time (IVRT). Both systolic and diastolic velocities using TDI have been shown to be reduced in aPH, including the subgroup of infants with CDH.96 Left ventricular dysfunction in aPH may be the primary pathology in the setting of post-capillary aPH. More commonly, LV dysfunction is secondary to a combination of RV impairment, due to ventricular interdependence and/or altered ventricular-ventricular interactions, reduced PBF with decreased LV preload, and myocardial ischemia with low coronary perfusion pressure. Decreased LV performance may contribute to the pathophysiology and clinical features of aPH. Decreased LV size and output have been shown to correlate with a requirement for advanced therapies in aPH, such as ECMO and prolonged mechanical ventilation.97 A low LV output (LVO) and diminished LV stroke volume with a normal or mildly reduced LV-EF due to right-to-left shunting (reduced preload) and changes in septal configuration are common findings in term and late preterm infants with resistance-driven aPH.76 The role of the LV in the pathophysiology of cardiorespiratory compromise in CDH is increasingly recognized,98,99 and early LV dysfunction and severe aPH are independent predictors of adverse outcome in “low risk” infants with the condition.100 LV dysfunction in CDH may result in post-capillary aPH secondary to pulmonary venous hypertension coexisting with classical, resistance-driven aPH. Afterload reduction with selective pulmonary vasodilators (e.g., iNO) in this context may worsen LV diastolic dysfunction and perpetuate hypoxemia. Determining the relative contribution of LV disease to symptomatology is important for guiding therapy, with inotropic (and lusitropic) support likely to be advantageous where LV impairment is significant, particularly where there is existing diastolic dysfunction. Prolongation of IVRT, reduction in the ratio of passive to active transmitral flow (E:A ratio; due to reliance of the stiff LV on the atrial “kick” for filling), and elevation in LV e:e′ and LV dilatation are suggestive of LV diastolic impairment. Echocardiography assessment of LV function is reviewed in detail in Chapters 9–11. LV output (LVO) measurement is suggested as a surrogate marker of systemic perfusion and LV function in aPH.1 The myocardial performance index (MPI) is generally used to estimate global LV function, although RV function in neonates may also be assessed using MPI72,76 and neonatal normative values have been published.79 Ventricular dysfunction leads to elevated MPI though increased isovolumic contraction and relaxation times, though, like many echocardiography measures, the MPI is load dependent. Both RV and LV MPI have been shown to be elevated in infants with aPH.101 Additional measures, such as ejection fraction and TDI of the IVS and LV free wall or STE, are also recommended for comprehensive appraisal of LV function.76 Assessment of extrapulmonary shunts is important for understanding PH hemodynamics and clinical decision-making. Right-to-left shunting at ductal and atrial level have been associated with adverse outcomes in heterogeneous groups of neonates with PH.87,102 It is important to recognize that the magnitude of the pulmonary-systemic shunt is a marker of disease severity rather than the cause of adverse outcomes and, in many patients, plays a beneficial role. For example, right-to-left ductal shunting both augments systemic perfusion and provides a pop-off mechanism for the RV exposed to high afterload. Although right-to-left atrial shunting is a hallmark of aPH due to high PVR, exclusive right-to-left shunting should prompt cardiac anatomical assessment to exclude total anomalous pulmonary venous drainage, in which there is an obligate right-to-left atrial shunt. Left-to-right flow via the atrial communication in the setting of markers of raised mPAP should raise concern regarding LV function and the presence of pulmonary venous hypertension and elevated left atrial pressure. It is in this subset of patients that pulmonary vasodilation is likely to be deleterious. Infants with congenital diaphragmatic hernia, where LV maldevelopment and dysfunction occur in conjunction with abnormalities of the pulmonary vascular bed, may present with this phenotype. With respect to ductal shunt, right-to-left flow for ≥30% of the cardiac cycle is consistent with raised mPAP, and right-to-left flow for >60% of systole has been associated with suprasystemic level PAP in term neonates with aPH.103 The neonatal RV myocardium is highly sensitive to increases in afterload, more so even than the LV.25 Right-to-left ductal shunting may offload the failing RV and support systemic perfusion, but at the expense of post-ductal oxygenation.15 Nonetheless, maintaining patency of the ductus with a low-dose prostaglandin infusion represents a therapeutic option in infants with severe aPH, impaired RV performance, and a restrictive PDA with the classic arterial aPH phenotype or in association with congenital diaphragmatic hernia. This effect may be magnified in patients with a large preductal systemic to venous connections, where post-ductal perfusion may be severely compromised; therefore maintaining patency of the ductus may be necessary to avert critical post-ductal hypoperfusion. Therapeutic approaches to aPH should be based upon a clear understanding of the physiologic concepts underpinning neonatal cardiovascular care and a detailed assessment of individual hemodynamics. Treatment goals include reduction of PVR, augmentation of RV performance as required, restoration of adequate systemic blood flow, ensuring adequate RV perfusion pressure, supportive care of the underlying condition, and correction of associated metabolic and hematologic derangements. A suggested algorithm for cardiovascular management in aPH is outlined in Figure 27.2. Optimizing lung recruitment guided by the pathophysiology of aPH and clinical, biochemical, and radiological response to treatment is important in terms of both supportive care for the underlying disease process, and for providing an optimal milieu for lowering PVR. Oxygen is a potent and selective pulmonary vasodilator; thus increased oxygen tension results in a reduction of PVR. Ideal oxygen saturation and/or arterial PO2 targets in PH are not completely clear, however, and there is wide practice variation.104 Pulmonary vasoconstriction occurs in response to both alveolar hypoxia and hypoxemia,105 though hyperoxia is also harmful106 and may blunt the vasodilatory response to iNO.107 A steep inverse relationship has been demonstrated between PAP and PaO2 in animal models between 20 and 40 mmHg.108 A plateau observed at 50 mmHg suggests that additional benefit in reduction of PVR is unlikely to be achieved with further increases in PaO2, and hyperoxia brings with it risks of free radical accumulation106 and an adverse effect on PVR.109 In general, oxygen saturations of 91–95% and PaO2 values between 50 and 80 mmHg are recommended.110 The optimal post-ductal PaO2, which is influenced by right-to-left ductal shunt, is unclear, however, and caution should be applied in placing strong emphasis on PaO2 values obtained from an umbilical arterial catheter in this setting.
Chapter 27: Pathophysiologically based management of pulmonary hypertension of the newborn
Introduction
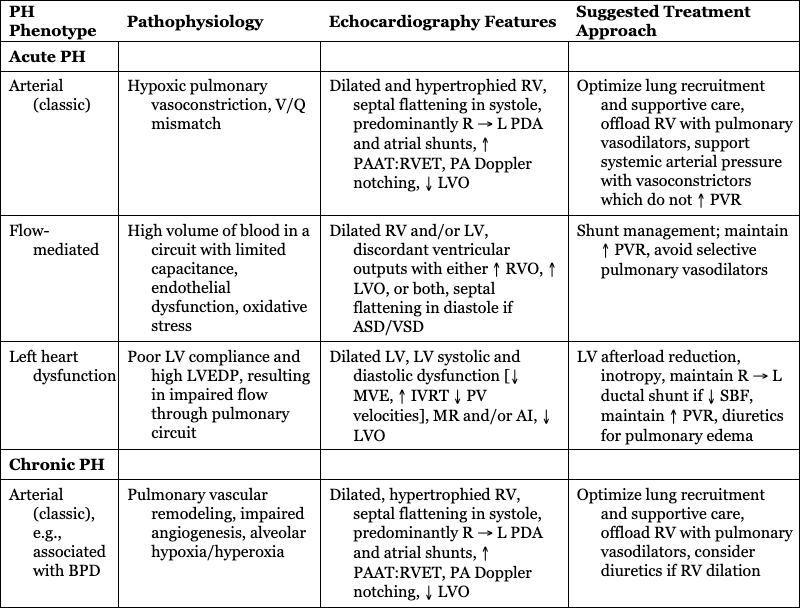
PH Phenotype
Pathophysiology
Echocardiography Features
Suggested Treatment Approach
Acute PH
Arterial (classic)
Hypoxic pulmonary vasoconstriction, V/Q mismatch
Dilated and hypertrophied RV, septal flattening in systole, predominantly R → L PDA and atrial shunts, ↑ PAAT:RVET, PA Doppler notching, ↓ LVO
Optimize lung recruitment and supportive care, offload RV with pulmonary vasodilators, support systemic arterial pressure with vasoconstrictors which do not ↑ PVR
Flow-mediated
High volume of blood in a circuit with limited capacitance, endothelial dysfunction, oxidative stress
Dilated RV and/or LV, discordant ventricular outputs with either ↑ RVO, ↑ LVO, or both, septal flattening in diastole if ASD/VSD
Shunt management; maintain ↑ PVR, avoid selective pulmonary vasodilators
Left heart dysfunction
Poor LV compliance and high LVEDP, resulting in impaired flow through pulmonary circuit
Dilated LV, LV systolic and diastolic dysfunction [↓ MVE, ↑ IVRT ↓ PV velocities], MR and/or AI, ↓ LVO
LV afterload reduction, inotropy, maintain R → L ductal shunt if ↓ SBF, maintain ↑ PVR, diuretics for pulmonary edema
Chronic PH
Arterial (classic), e.g., associated with BPD
Pulmonary vascular remodeling, impaired angiogenesis, alveolar hypoxia/hyperoxia
Dilated, hypertrophied RV, septal flattening in systole, predominantly R → L PDA and atrial shunts, ↑ PAAT:RVET, PA Doppler notching, ↓ LVO
Optimize lung recruitment and supportive care, offload RV with pulmonary vasodilators, consider diuretics if RV dilation
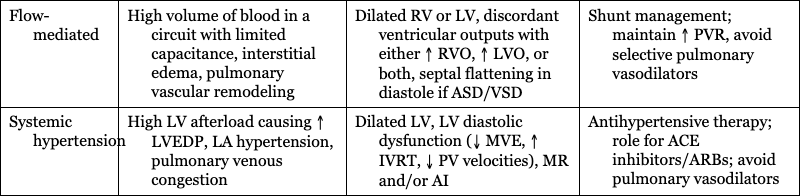
Flow-mediated
High volume of blood in a circuit with limited capacitance, interstitial edema, pulmonary vascular remodeling
Dilated RV or LV, discordant ventricular outputs with either ↑ RVO, ↑ LVO, or both, septal flattening in diastole if ASD/VSD
Shunt management; maintain ↑ PVR, avoid selective pulmonary vasodilators
Systemic hypertension
High LV afterload causing ↑ LVEDP, LA hypertension, pulmonary venous congestion
Dilated LV, LV diastolic dysfunction (↓ MVE, ↑ IVRT, ↓ PV velocities), MR and/or AI
Antihypertensive therapy; role for ACE inhibitors/ARBs; avoid pulmonary vasodilators
Clinical presentation
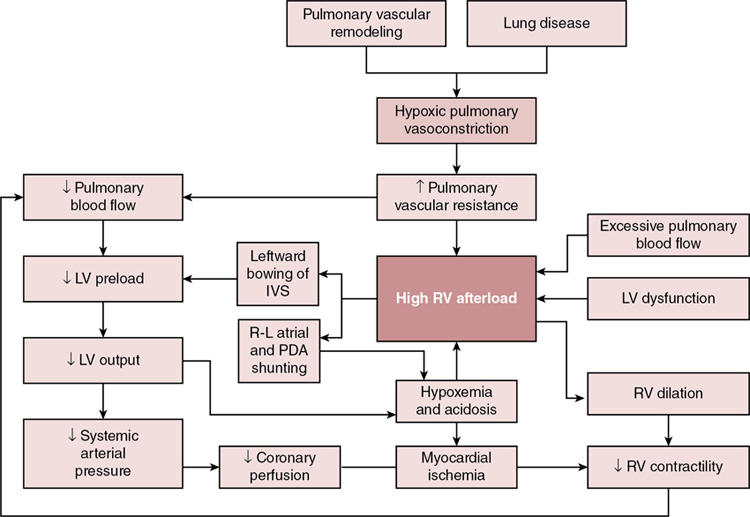
Pathophysiology
Acute pulmonary hypertension of arterial (classic) origin
Acute pulmonary hypertension related to excessive pulmonary blood flow
Acute pulmonary hypertension secondary to left ventricular dysfunction
Chronic pulmonary hypertension (cPH)
Chronic pulmonary hypertension secondary to BPD
Chronic pulmonary hypertension secondary to excessive pulmonary blood flow
Chronic pulmonary hypertension secondary to LV dysfunction
Delineation of PH phenotype
Echocardiography-based assessment in aPH
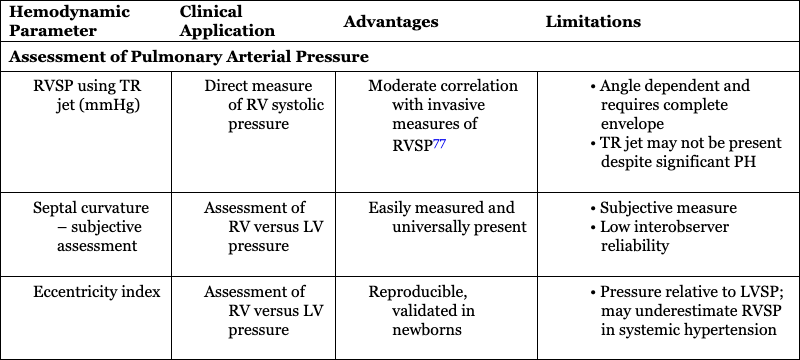
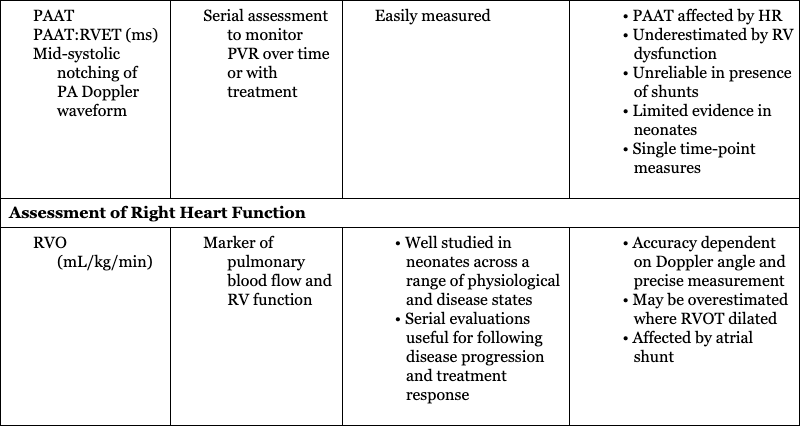


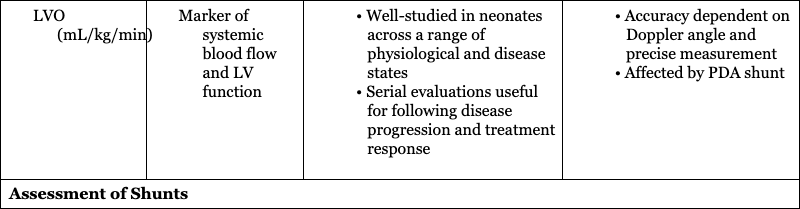
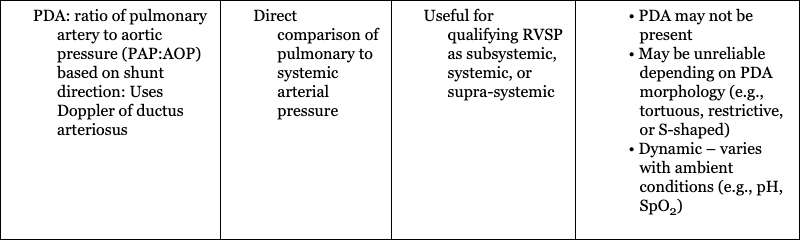
Hemodynamic Parameter
Clinical Application
Advantages
Limitations
Assessment of Pulmonary Arterial Pressure
Assessment of Right Heart Function
Assessment of Systemic Blood Flow
Assessment of Shunts
Evaluation of pulmonary artery pressure and pulmonary vascular resistance
Assessment of right ventricular function
Assessment of left ventricular function
Assessment of extrapulmonary shunts
Treatment principles
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Obgyn Key
Fastest Obstetric, Gynecology and Pediatric Insight Engine