DEFINITION
The complaint of pallor indicates a perceived decrease in rubor in the skin and mucous membranes of a child, which is associated with decreased oxyhemoglobin delivery to the skin or mucous membranes. Potential causes include decreased blood flow, which may be regional (e.g., thrombosis) or systemic (e.g., shock), and normal blood flow with decreased oxygen-carrying capacity (e.g., anemia).
In most cases, the finding of pallor demands that anemia first be considered as this is the most common cause. Exceptions include children who have a constitutional cause of pallor due to their fair complexion and lack of exposure to sunlight. However, most children with pallor should be considered to have low hemoglobin, which should be measured. Ordinarily, a complete blood count with differential count, red blood cell (RBC) indices, and reticulocyte count guide the clinician in differentiating the many causes of anemia and in determining unusual situations in which pallor is not related to anemia. In addition, a peripheral smear with examination of RBC morphology may further guide the clinician in determining the etiology of anemia.
CAUSE AND FREQUENCY
Pallor may be divided into causes involving normal hemoglobin (Table 10-1) and those involving a low hemoglobin level. The etiology of anemia may be separated into causes due to decreased RBC production, increased RBC destruction, or acute blood loss. Causes due to decreased RBC production are generally associated with low reticulocyte count while causes due to increased RBC destruction or acute blood loss are generally associated with increased reticulocyte count. Anemia may also be considered in relation to mechanism, such as trauma, toxin, metabolic tumor, congenital, or mixed etiology (Table 10-2), or in relation to RBC morphology (Table 10-3).
TABLE 10-1. Pallor with anemia.
Decreased erythrocyte or hemoglobin production
Increased Erythrocyte Destruction
Blood Loss
aCan present as life-threatening emergencies
TABLE 10-2. Etiology of anemia by mechanism (common causes of pallor).
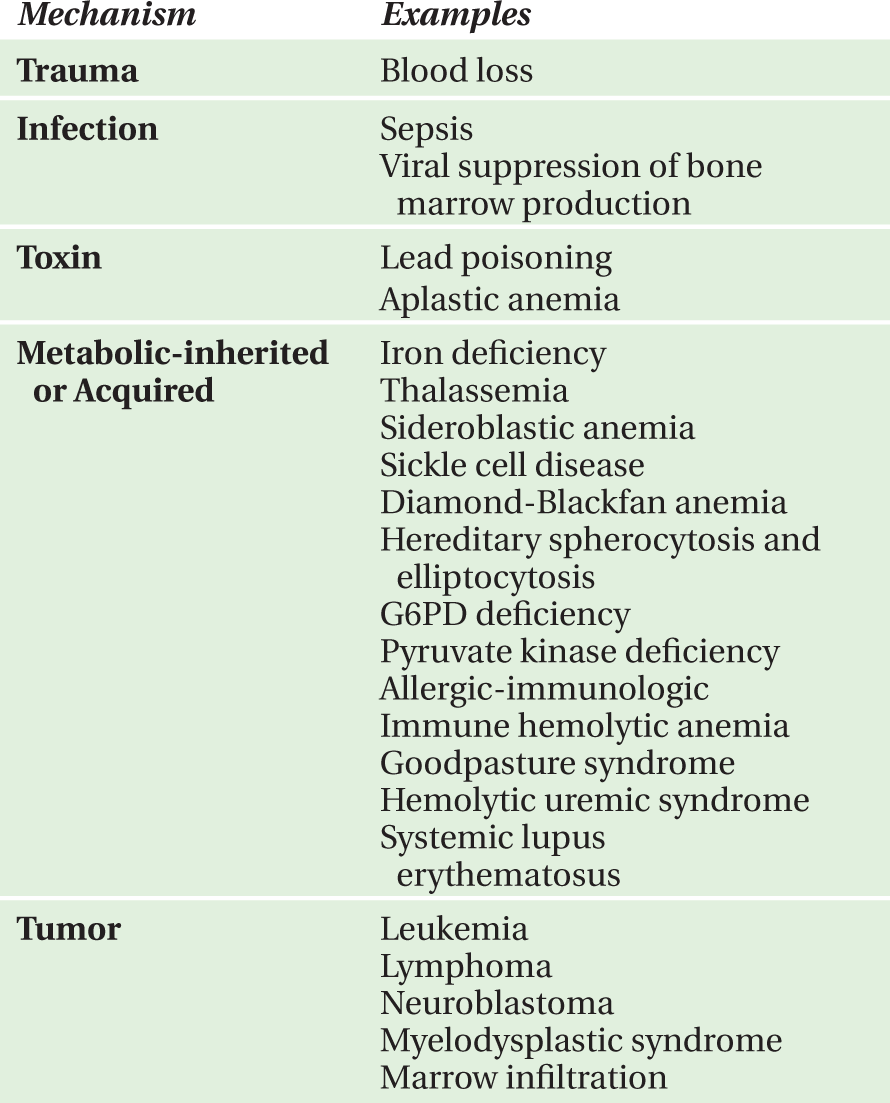
TABLE 10-3. Etiology of anemia by red blood cell morphology.
Microcytic Anemia
Normocytic Anemia
Macrocytic Anemia
QUESTIONS TO ASK AND WHY
• Is there hemodynamic instability or need for emergent intervention?
—The most important question to ask is whether a child’s pallor is secondary to severe anemia with hemodynamic compromise that requires emergent intervention and stabilization with fluid resuscitation until transfusion of packed RBCs. This question also helps to determine whether severe anemia is secondary to acute external hemorrhage that may require immediate surgical intervention. One can also elucidate if a child’s pallor is secondary to decreased blood flow from acute thrombosis or systemic shock further guiding therapy.
• What are the child’s dietary habits?
—Because the most common cause of pallor is iron deficiency anemia, important clarifying questions pertain to diet. Diets containing large quantities of cow’s milk may raise concern for iron deficiency due to lack of iron intake and loss of appetite for iron-rich foods.
aCan present as life-threatening emergencies
• Is there a family history of anemia, splenectomy, or gallbladder surgery?
—Many RBC membrane disorders are inherited. Some disorders, such as hereditary spherocytosis and elliptocytosis, frequently necessitate splenectomy due to RBC sequestration. Inherited diseases causing hemolysis, such as sickle cell disease, may be recognized in some families by a history of gallbladder surgery for gallstones.
• Are there signs of systemic illness, fatigue, growth failure, weight loss, or lymphadenopathy?
—The presence of these signs or symptoms should raise concern for malignancy.
• Was there exposure to certain medications or toxins?
—Questions regarding exposure to toxins or medications should be asked as these may cause anemia by hemolysis (e.g., sulfonamide or nitrofurantoin exposure in a patient with glucose- 6-phosphate dehydrogenase [G6PD deficiency]) or by suppressing bone marrow production of RBCs (e.g., trimethoprim/sulfamethoxazole).
• Did pallor develop quickly or gradually?
— Information about the progression of anemia provides clues about the cause. Gradual onset suggests iron deficiency while acute onset with jaundice suggests hemolysis. In cases of gradual development of pallor, additional questions should focus on sources of iron loss or blood loss, such as history of heavy menstruation, blood in stool or melena, or multiple blood draws for diagnostic testing.
SUGGESTED READINGS
1. Poncz, M. Pallor. In: Schwartz MW, ed. Pediatric Primary Care: A Problem-Oriented Approach. 2nd ed. Chicago, London, Boca Raton, Littleton, MA: Year Book Medical Publishers; 1987.
2. Segel GB, Hirsh MG, Feig SA. Managing anemia in pediatric office practice: Part I. Pediatr Rev. 2002;23:75-84.
3. Shah S. Pallor. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2010.
4. Bizzaro M, Colson E, Ehrenkranz R. Differential diagnosis and management of anemia in the newborn. Pediatr Clin North Am. 2004;51:1087-1107.
CASE 10-1
Three-Week-Old Boy
HISTORY OF PRESENT ILLNESS
The patient was a 3-week-old Caucasian male, born at 38 weeks gestation, who presented to an outpatient clinic for evaluation of anemia. Shortly after birth, he was noted to be pale in the newborn nursery. At that time, his hemoglobin was 12.3 g/dL with a mean corpuscular volume (MCV) of 120 fL. The hemoglobin was repeated at 2 weeks of age and was 8.1 g/dL with a reticulocyte count of 1.2%. He had initial problems with weight gain that improved after his mother started pumping and giving him breast milk via bottle in addition to breastfeeding. His parents thought he was often “sleepy” and were specifically concerned that he frequently fell asleep during feeds. He had no vomiting, diarrhea, fever, or cough. He had normal gold-colored bowel movements. There had been no change in urine color and he had no rash.
MEDICAL HISTORY
The infant was a twin born via vaginal delivery at 38 weeks gestational age following in vitro fertilization. He had transverse lie and was delivered vertex after external manipulation. The mother had anemia during pregnancy and her only medication was prenatal vitamins. Her blood type was O positive. The birth weight was 2470 g; the infant’s twin sibling weighed 2900 g at birth. On his first day of life, the infant was noted to have a swollen right upper leg with significant bruising. The initial radiograph was normal but a subsequent film showed evidence of a healing fracture that was presumably related to birth trauma. There was no history of jaundice in the newborn nursery.
The infant received multivitamin with iron. He did not have any known allergies. He received a diet of breast milk with occasional formula supplementation.
The family history was remarkable for the mother’s anemia, which did not require treatment. The maternal grandmother also had a history of anemia. Both the maternal grandmother and aunt required cholecystectomy for gallstones.
PHYSICAL EXAMINATION
T 36.7°C; HR 166-230 bpm; RR 46-66/min; BP 70/37 mmHg
Weight 3.1 kg (25th percentile); Height 50 cm (50th percentile); Head circumference 36 cm (~75th percentile)
On examination, the infant was remarkably pale appearing, awakened easily, and cried. The anterior fontanel was open and flat. The conjunctivae were pale and the sclerae were anicteric. Mucous membranes were moist. The clavicles were intact. The infant was tachypneic but the lungs were clear to auscultation. On cardiac examination, the patient had a normal S1 and S2 and there was a 3/6 systolic murmur best appreciated at the left upper sternal border but not heard along the back. There were no gallops or rubs. The liver edge was palpable but the spleen was not. On musculoskeletal examination, the site of known extremity fracture had minimal edema but no tenderness and the right distal femur appeared larger than the left. The remainder of the examination was normal.
DIAGNOSTIC STUDIES
Complete blood count revealed the following: hemoglobin, 3.9 g/dL; 11 300 WBCs/mm3 (1% metamyelocytes, 43% segmented neutrophils, 34% lymphocytes, and 19% monocytes); 430 000 platelets/mm3; MCV, 117 fL; red cell distribution width, 17; and reticulocyte count, 0.3%. The peripheral blood smear revealed a few small spherocytes but no schistocytes, burr cells, or target cells.
COURSE OF ILLNESS
The infant was admitted for supportive care and a diagnostic evaluation begun. What are the likely cause for his anemia?
DISCUSSION CASE 10-1
DIFFERENTIAL DIAGNOSIS
Table 10-4 lists the differential diagnosis of anemia in an infant. This patient presented with pallor during infancy and no history of jaundice, which lessened the likelihood of hemolysis. There was no concern of a dietary etiology because of the baby’s young age, there was no chronic illness apparent, and no history of blood loss; therefore, the focus shifted to a congenital defect in red cell production. There was a remarkable drop in the hemoglobin along with a very poor bone marrow response as far as production of reticulocyte. The white cells and platelets were normal and the anemia was macrocytic. The sum of these findings indicated a defect of red cell production. There was also the physical examination finding of a possible skeletal anomaly in the distal right femur.
Red cell aplasia may be congenital or acquired. Most of the acquired forms occur during adulthood or adolescence. In childhood the major causes of red cell aplasia are Diamond-Blackfan anemia, transient erythroblastopenia of childhood, and acquired aplasia of red cells associated with chronic hemolysis (as seen in sickle cell disease).
In this case, there was no evidence of acute or chronic hemolysis and the patient was too young to be considered for transient erythroblastopenia of childhood. Another possible etiology to be considered was Fanconi anemia. Fanconi anemia is an autosomal recessive disorder associated with aplastic anemia, short stature, skeletal defects, pigmentation changes, and other abnormalities. Some cases of Fanconi anemia are diagnosed in the first year of life. The anemia involves all cell lines and the diagnosis is established via bone marrow analysis and genetic studies. However, the patient had anemia without abnormalities in the white blood cell or platelet count, thus Diamond-Blackfan was the most plausible diagnosis.
DIAGNOSIS
The infant was admitted with the diagnosis of anemia secondary to a hypoproductive state. The infant was felt to have symptomatic anemia (difficulty feeding); therefore, he was transfused with a total of 15 mL/kg of packed red blood cells administered in three separate aliquots. He underwent bone marrow aspiration, which revealed absence of red cell precursors with normal granulocyte precursors, a finding consistent with Diamond-Blackfan anemia. Following the packed red cell transfusion, the infant’s hemoglobin was 8.7 g/dL. He was more active and was no longer in respiratory distress. The baby received corticosteroids at 2 mg/kg/day, and was given an additional 5 mL/kg of packed red blood cells prior to discharge. The final diagnosis in this case was Diamond-Blackfan anemia.
INCIDENCE AND EPIDEMIOLOGY OF DIAMOND-BLACKFAN ANEMIA
The precise incidence is unknown; however, it is estimated that there are 300-1000 new cases of red cell aplasia annually in the United States. Diamond-Blackfan occurs primarily in infancy. Studies show that 10% of patients are anemic at birth, 25% by 1 month, 50% by 3 months, and 70% by 1 year. It is seen in all ethnic groups but primarily in Caucasians. There is no gender predominance.
CLINICAL PRESENTATION OF DIAMOND-BLACKFAN ANEMIA
Pallor caused by anemia in the early months of life characterizes this form of anemia. About one-third of patients have at least one associated finding, including characteristic facies, thumb anomalies, short stature, eye abnormalities including glaucoma, renal anomalies, hypogonads, skeletal anomalies, congenital heart disease, and mental retardation. There is a wide range of involvement. Some patients progress to full aplastic anemia and about 5% may develop leukemia or myelodysplasia.
DIAGNOSTIC APPROACHES
Complete blood count and peripheral blood smear. The mean hemoglobin level for all patients with Diamond-Blackfan anemia is 7 g/dL at diagnosis. Infants diagnosed in the first 4 months of life typically have hemoglobin levels of 4 g/dL at presentation. The reticulocyte count is decreased or zero. The peripheral blood smear reveals macrocytes, anisocytosis, and teardrops. The WBC counts are normal in most patients; though in 20%, WBC counts are less than 5000/mm3.
Bone marrow aspirate. Bone marrow aspirates and biopsies show normal cellularity, myeloid cells, and megakaryocytes. Approximately 90% of patients have erythroid hypoplasia or aplasia. The remaining 10% of patients have either normal erythroblast number and maturation or erythroid hyperplasia with maturation arrest. Despite the variable marrow findings, all patients have reticulocytopenia, indicating some form of ineffective erythropoiesis and delayed precursor maturation.
Other studies. Serum iron, ferritin, folate, vitamin B12, and erythropoietin are all elevated. In most cases, there is an increase in erythrocyte adenosine deaminase (eADA). Among patients with bone marrow failure syndromes, eADA had a sensitivity of 84%, specificity of 95%, and positive and negative predictive values of 91% for the diagnosis of Diamond-Blackfan anemia. Genetic studies have shown one site on chromosome 19 and other gene defects have also been associated.
TREATMENT
The treatment includes use of corticosteroids. The current recommended dose is 2 mg/kg/day of prednisone, administered in 3-4 divided doses. Reticulocytes appear within 1-2 weeks but the rise in hemoglobin is delayed for several more weeks. Once the hemoglobin level reaches 10 g/dL, the steroid dose is gradually tapered until the patient receives a single daily dose that adequately maintains appropriate hemoglobin levels. Response followed by steroid dependence is seen in 60% of patients. Approximately one-fifth of the steroid-responsive patients may ultimately be maintained without steroids.
Approximately 30%-40% of patients have poor or no response to steroids and require chronic transfusion therapy to maintain normal hemoglobin. These children require leukocyte-depleted packed red blood cell transfusions every 3-6 weeks with the goal of keeping the hemoglobin level greater than 6 g/dL. Concurrent chelation of iron with subcutaneously administered desferrioxamine may decrease some chronic transfusion-related complications. Complications of chronic transfusion therapy are similar to other conditions that employ this modality (e.g., thalassemia). Bone marrow transplantation has been successful for some patients.
Median survival is 43 years of age but approximately 13% of patients die within the first six years of life. Deaths occur from complications of iron overload, pneumonia, sepsis, and occasionally from transplant-related complications, leukemia, and pulmonary emboli.
SUGGESTED READINGS
1. Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT, eds. Nathan and Oski’s Hematology of Infancy and Childhood. 6th ed. Philadelphia: Saunders; 2003:280-365.
2. Ball SE, McGuckin CP, Jenkins G, et al. Diamond- Blackfan anaemia in the UK: analysis of 80 cases from a 20 year birth cohort. Br J Haematol. 1996;94:645-653.
3. Willing TN, Draptchinskaia N, Dianzani I, et al. Mutations in ribosomal protein S19 gene and Diamond Blackfan anaemia: wide variations in phenotypic expression. Blood. 1999;94:4294-4306.
4. Willing TN, Gazda H, Sieff CA. Diamond Blackfan anaemia. Curr Opin Haematol. 2000;7:85-94.
5. Fargo JH, Kratz CP, Giri N, et al. Erythrocyte adenosine deaminase: diagnostic value for Diamond-Blackfan anemia. Br J Haematol. 2012 [Epub ahead of print, PMID 23252420].
Twelve-Month-Old Girl
HISTORY OF PRESENT ILLNESS
A 12-month-old Caucasian girl presented to the emergency department with pallor. On the day of arrival, the grandparents had arrived from Florida and were alarmed at her appearance (Figure 10-1) prompting a visit to the emergency department. The child had last been seen by her grandparents a few months prior and at that time she appeared well. The parents conceded that she did appear more pale than usual. There was no fever, rash, vomiting, or diarrhea and her activity level was normal. She had no jaundice and had not traveled recently, although the parents had visited Puerto Rico 2 weeks ago.
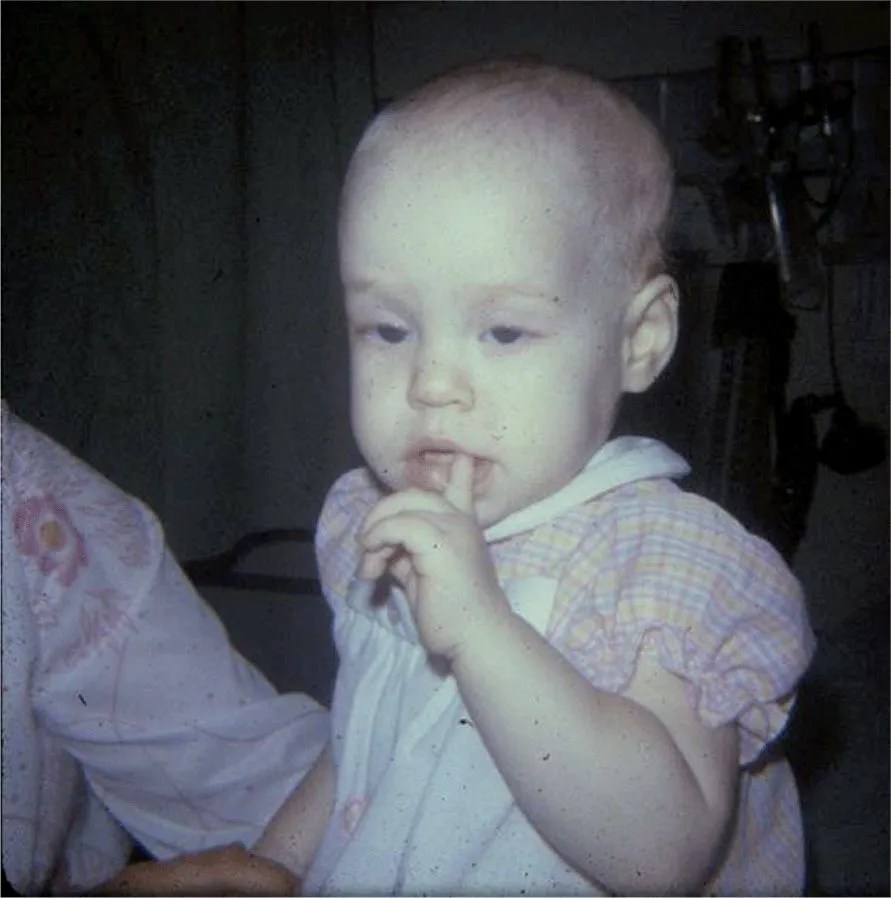
FIGURE 10-1. Photograph of the child showing marked pallor.
MEDICAL HISTORY
The birth history was unremarkable. She had a febrile illness at 6 months of age but did not require hospitalization. Evaluation at that time included a complete blood count (see Diagnostic Studies) and a blood culture positive for Staphylococcus epidermidis, which was felt to be a contaminant. Her immunizations were up-to-date. She was breast fed until 6 months of age after which she began drinking whole milk. She was a finicky eater but had been growing well. There were no pets in the home and her development was normal.
PHYSICAL EXAMINATION
T 36.1°C; HR 145 bpm; RR 38/min; BP 97/53 mmHg; SpO2 99% in room air
The girl was pale but alert and playful. The conjunctivae were also pale but without injection or discharge. There was no lymphadenopathy and her neck was supple. A 2/6 systolic murmur was heard at the left upper sternal border without radiation. The lungs were clear to auscultation. On abdomen examination, there was no splenomegaly or hepatomegaly and there were no rash or petechiae on skin.
DIAGNOSTIC STUDIES
At 6 months of age, the complete blood count revealed the following: WBCs, 15 200/mm3 (71% segmented neutrophils, 25% lymphocytes, and 4% monocytes); hemoglobin, 11.2 g/dL; 365 000 platelets/mm3, with an MCV of 78 fL and an RDW was 17.3.
Her current studies revealed the following: WBCs, 8300/mm3 (58% segmented neutrophils, 31% lymphocytes, and 11% monocytes); hemoglobin, 3.4 g/dL; and 410 000 platelets/mm3 with an MCV 59 fL and an RDW of 15.1. The reticulocyte count was 1.4%. Stool was hemoccult negative.
COURSE OF ILLNESS
The history, examination (Figure 10-1), and laboratory findings suggested a diagnosis that was subsequently confirmed.
DISCUSSION CASE 10-2
DIFFERENTIAL DIAGNOSIS
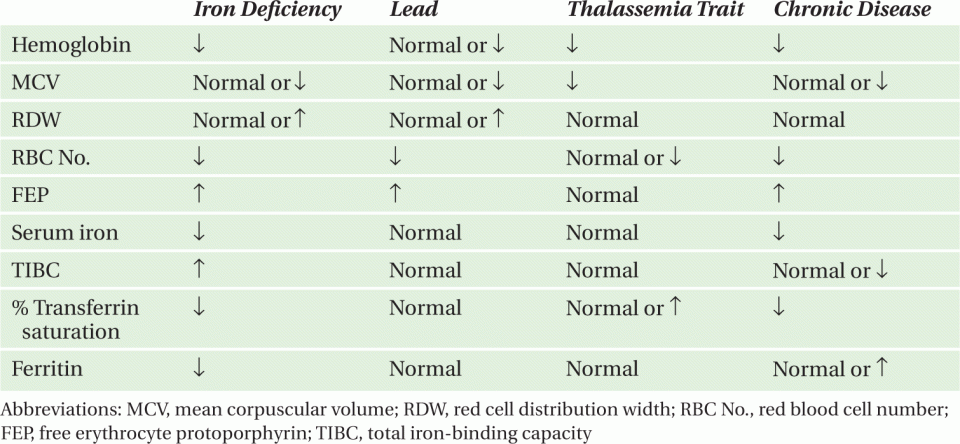
Table 10-5 lists the differential diagnosis of micro-cytic anemia in children. Causes of iron deficiency include poor bioavailability, decreased iron absorption, blood loss, insufficient intake, and disruption of enteric mucosa or loss of functional bowel. Alkaline gastric pH reduces the solubility of inorganic iron, impeding absorption. Chronic use of acid pump blockers, vagotomy (for severe gastroesophageal reflux), and impaired gastric parietal cell function in pernicious anemia may compromise iron absorption. Iron absorption may also be disrupted after surgical bowel resection often following volvulus or intussusception. Iron deficiency in such cases develops slowly and may not become evident for several years. Blood loss is a leading cause of iron deficiency. Common causes of gastrointestinal blood loss in children include Meckel diverticulum, cow milk protein allergy, and parasitic infestation; however, blood loss from hematuria or pulmonary hemorrhage can also occur. In this case, the dietary history suggested a likely cause.
TABLE 10-5. Evaluation of microcytic anemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


