FIGURE 17.1 Cardiac action potential.
No set panel of laboratory tests should be ordered for every case of potential poisoning. Rather, one should tailor studies to assess for proof of exposure, to uncover possible complications, or for prognostic reasons. However, in the case of the child ill from an unknown poison, evaluation of the basic metabolic panel often provides a useful first step toward narrowing the differential diagnosis. First, electrolyte abnormalities commonly associated with certain exposures might suggest a diagnosis in the proper clinical setting. For example, altered mental status with hyponatremia commonly results from binging with MDMA (3,4-methylenedioxymethamphetamine or “ecstasy”), which causes the syndrome of inappropriate antidiuretic hormone release (SIADH) (7,8).
Additionally, calculation of an anion gap (AG) helps distinguish common toxicologic causes of a metabolic acidosis. Patients with a metabolic acidosis in whom the AG (Na-[Cl+HCO3]) is elevated (> 12 mmol/L) have an unmeasured anion present in their serum. A mnemonic to remember the potential causes of an elevated AG is KULTS: ketoacids, uremia, lactic acid, toxic alcohols, and salicylates. Ketones often result from an alcoholic or a starvation ketoacidosis. Uremia results from any toxin that causes renal insufficiency, and the diagnosis is confirmed by finding a markedly elevated blood urea nitrogen. Metabolic acidosis with an elevated lactate concentration accounts for most cases of toxin-induced elevated AG metabolic acidosis and etiologies range from cellular asphyxiants, such as cyanide, to antidiabetics, such as metformin. Toxic alcohols, such as ethylene glycol, undergo metabolism to an organic acid. In the case of ethylene glycol, the metabolites glycolic acid and oxalic acid result in an AG metabolic acidosis and renal failure may occur. Salicylate derivatives, such as aspirin or oil of wintergreen (methyl salicylate), cause both an AG metabolic acidosis and a primary respiratory alkalosis.
■ SYNDROMIC APPROACH TO POISONED PATIENTS
Given the vast array of potential poisons, the complex clinical findings they produce, and the limitation of the analytical laboratory in identifying specific toxins in real time, the ability to discern the nature of a toxic exposure on clinical grounds becomes critical. However, through recognition of toxicological syndromes, or “toxidromes,” a clinician can identify the largest threats to a patient’s life and institute empiric therapy while additional diagnostic strategies continue. The following paragraphs are intended to provide a framework with which to consider common poisonings and to highlight the related critical interventions and potential antidotes.
Altered Mental Status
While “altered mental status” frequently constitutes the entirety of a documented chief complaint, it fails to distinguish between the distinct clinical entities such as sedation, agitation, confusion, and hallucinations. Within these descriptors are a range of etiologies and severities with differing implications. For example, depressed mental status may range from sleepiness to coma, and this is often accompanied by progressive respiratory depression. An agitated patient, on the other hand, may present with a subtle delirium or an obvious psychosis. Some of these patients may be a danger to themselves or others and require physical and chemical restraint. Implicit in the described management strategies is that the fundamentals are addressed for each patient. These include acting on cardiorespiratory vital sign abnormalities, core body temperature, and rapid bedside testing such as glucometry and ECG.
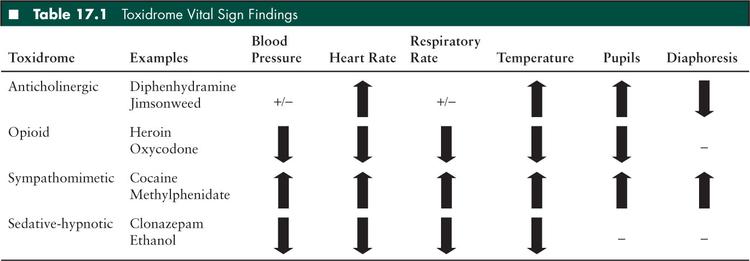
Opioid Toxidrome
While most commonly noted for its associated “pinpoint pupils,” decreased ventilation resulting from both decreased respiratory rate and decreased tidal volume remain the most consequential effects of this poisoning (Table 17.1). Implicated drugs are agonists at the opioid μ receptor in the CNS and include oxycodone and heroin. Due to the contemporary ubiquity of these analgesics, unintentional poisonings in children have reached epidemic levels (9). Other effects include mild hypotension and bradycardia but these findings are typically less concerning than the respiratory effects.
The primary intervention in an opioid poisoned child is to provide ventilatory support. Naloxone, an opioid antagonist, may be administered to reverse the clinical effects of an opioid agonist (Table 17.2). The usual pediatric starting dose of naloxone in an opioid naive child is 0.1 mg/kg IV, with a first dose maximum of 2 mg. This is paradoxically higher than the dose typically recommended for adults, but most children who are exposed to opioids are indeed opioid naive and thus not at risk of opioid withdrawal following naloxone treatment. In addition, often the offending agent is a synthetic opioid or semisynthetic opiate derivative that may require higher dosing for reversal. In children or adolescents who are believed at risk for opioid dependence, the starting dose should be 0.001 mg/kg (maximum 0.04 mg). These starting doses can be administered and swiftly escalated as needed to as much as 2 to 4 mg in toddlers and 8 to 10 mg in adolescents if opioid toxicity is suspected or confirmed. Buprenorphine, a recently popularized partial-agonist at the opioid μ receptor, may be particularly resistant to reversal by naloxone (10–12). If the patient requires more than 1 dose of naloxone or the drug effect is expected to outlast the antidote effect, a naloxone infusion may be preferred, with a starting rate of two-thirds of the effective dose infused per hour, titrated to normal respiratory rate.

Sedative-Hypnotic Toxidrome
Patients poisoned by sedative hypnotics present with coma, generally associated with normal vital signs. Common causes of this relatively benign toxidrome include benzodiazepines, such as diazepam or clonazepam, newer sleep agents, such as zolpidem or eszopiclone, and ethanol. All of these enhance inhibitory tone in the CNS through GABA agonism. Most children exposed to one of these drugs in isolation tend to preserve their respiratory drive and hemodynamics. On the other hand, patients with mixed exposures (e.g., benzodiazepine with ethanol) often require airway protection and other supportive care. Additionally, γ-hydroxybutyric acid is a popular club drug that results in the sedative-hypnotic toxidrome.
Most patients demonstrating the sedative-hypnotic toxidrome are managed expectantly and do not require any specific treatment. Flumazenil, a competitive antagonist at the benzodiazepine receptor, should only be administered to patients with acute benzodiazepine exposure who have absolutely no history of chronic benzodiazepine use. When given to patients with chronic exposure to benzodiazepines, it can precipitate the benzodiazepine withdrawal syndrome including status epilepticus (13). As a result, flumazenil is not generally recommended for benzodiazepine intoxication in adults or teenagers due to the inability to exclude a history of chronic use with certainty. In young children, however, the drug may be used diagnostically to confirm and treat benzodiazepine overdose as a suspected cause of altered mental status. The recommended starting dose of flumazenil is 0.01 mg/kg (max 0.2 mg) IV at 0.1 mg per minute. In this very specific treatment niche, flumazenil may save a child with a clear history of benzodiazepine naiveté from extensive testing such as a computerized tomography imaging of the head or a lumbar puncture, or unnecessary endotracheal intubation.
Agitated Delirium
Agitation poses unique challenges that are not encountered with other medical presentations. It requires that the provider take active measures to protect the patient with little opportunity to gather data or perform a thorough physical examination. Once these emergent interventions are implemented, close consideration of the patient’s clinical findings enables identification of a likely cause and the initiation of therapy.
Sympathomimetic Toxidrome
Causes of sympathomimetic effects range from use of illicit drugs, such as cocaine and ecstasy, to overdoses of commonly prescribed attention deficit hyperactivity disorder medications, such as methylphenidate and dextroamphetamine. By different mechanisms, drugs in this class are agonists at adrenergic receptors in the sympathetic nervous system. Vital sign abnormalities include tachycardia, hypertension, and hyperthermia. On physical examination, patients exhibit mydriatic pupils that are reactive to light, diaphoresis, and normal bowel sounds.
Management of these patients centers on controlling their agitation with benzodiazepines and promptly diagnosing and treating any vital sign abnormalities, particularly hyperthermia. Importantly, physical restraints, which are often needed initially, should serve only as a bridge to chemical restraint. Physical restraint without chemical sedation allows the patient to struggle and potentially develop hyperthermia, metabolic acidosis, and cardiac dysrhythmias (14,15). While various drugs have been used for sedation of the sympathomimetic patient, benzodiazepines offer the greatest safety profile and have the most logical pharmacologic mechanism (i.e., GABA agonism). As a result, the patient’s heart rate and blood pressure improve and seizures are prevented. Diazepam starting at 0.1 mg/kg IV or midazolam starting at 0.025 mg/kg IV or intramuscular (IM) (max 5 mg) is recommended to gain behavioral control of the agitated sympathomimetic patient. Haloperidol, a butyrophenone antipsychotic that functions by blockade of dopamine receptors, is commonly utilized as a chemical restraint agent. However, haloperidol is not the drug of choice in this patient population due to its tendency to lower the seizure threshold, its ability to cause hyperthermic syndromes, and its association with torsades de pointes.
Hyperthermia is a primary contributor to death in these patients and necessitates rapid cooling (16,17). In cases of mild temperature elevation, IV benzodiazepines to control agitation coupled with external measures such as fans and ice packs typically result in prompt cooling. In cases of severe hyperthermia (> 105°F), these simple interventions are often inadequate and ice water immersion becomes the intervention of choice. If unable to obtain a tub or cholera bed in which to submerge the patient, cooling in bed may be effected by circumferentially covering the patient with a sheet and packing it with ice directly on the entire trunk. One should move quickly to paralysis and endotracheal intubation along with ice water immersion if unable to rapidly cool an awake patient (18).
It is important to consider the patient’s particular complaints and the potential therapies within the context of their sympathomimetic toxidrome. For example, an otherwise healthy teenager who uses methamphetamine is at increased risk for subarachnoid hemorrhage, myocardial infarction, and also aortic, vertebral, and carotid dissection. While these disease processes classically spare the pediatric population, they must be considered in symptomatic patients with recent use of a sympathomimetic substance. Additionally, therapy must be guided to avoid unintended clinical effects; for example, β-blockers are contraindicated in patients with a sympathomimetic toxidrome. By blocking the β2 receptor in the coronary vasculature, they may lead to unopposed α1 receptor agonism causing worsened vasoconstriction and cardiac ischemia (19). Judicious treatment with IV benzodiazepines leads to well-controlled blood pressure and relief of chest pain in most cases.
Anticholinergic Toxidrome
More accurately called the antimuscarinic toxidrome, this syndrome results from any drug that is an antagonist to acetylcholine at muscarinic receptors. Common causative pharmaceuticals include diphenhydramine, tricyclic antidepressants (TCAs), and first generation antipsychotics such as haloperidol. Jimsonweed (Datura stramonium), a natural source of atropinelike chemicals can also cause this toxidrome (20–22). The agitation of the anticholinergic patient is classically milder than that of the sympathomimetic patient. They typically mumble quietly to themselves with a characteristic dysarthria often termed “cotton ball mouth” and appear to be hallucinating as they pick with their fingers into the air and on their bed sheets. Their physical examination can also be distinguished from that of the sympathomimetic patient by their lack of diaphoresis. Due to muscarinic blockade at salivary and sweat glands, these patients have dry mucous membranes as well as difficulty dissipating heat by sweating, resulting in cutaneous vasodilation and a flushed appearance of their skin. Loss of vagal tone results in tachycardia. Decreased cholinergic input results in dilated pupils that are poorly reactive to light. The classic prose taught to students highlights the pertinent physical findings of this toxidrome: “Red as a beet (flushed), hot as a hare (hyperthermic), mad as a hatter (hallucinating), dry as a bone (anhydrotic), and full as a flask (urinary retention).”
Initial management of the anticholinergic patient mirrors that of the sympathomimetic patient. A core body temperature identifies hyperthermic patients in need of rapid cooling. Agitation is managed with IV benzodiazepines and patients are otherwise supported until toxicity resolves. Alternatively, known or suspected anticholinergic patients who have normal ECGs can be safely reversed with the antidote physostigmine. This drug inhibits acetylcholinesterase in the neurosynaptic junction allowing the accumulation of acetylcholine and the ability to overcome competitive antagonism at the muscarinic receptor. The starting dose is 0.02 mg/kg (max 0.5 mg) infused over at least 5 minutes. Atropine should be at the bedside and administered if symptoms of cholinergic toxicity occur (i.e., bradycardia, hypersalivation.) It is contraindicated in patients with a wide QRS complex or who have taken a TCA (23). Anticholinergic patients treated with physostigmine tend to normalize their vital signs and mental status within minutes. This intervention may provide a definitive diagnosis in the altered child with an anticholinergic toxidrome, thereby alleviating the anxiety of family members and sparing the child further invasive testing.
Toxin-Induced Seizures
Toxins that enhance excitatory input or decrease inhibitory tone can lead to seizure. Glutamate is the most pertinent excitatory neurotransmitter in the brain and acts via agonism at the NMDA receptor and others. Ibotenic acid, for example, a poison found in the mushroom Amanita muscara is an NMDA receptor agonist, and causes neuroexcitation and seizures (24). Alternatively, tetramethylenedisulfotetramine, an illegal rodenticide colloquially known as “tetramine,” inhibits GABA in a noncompetitive fashion (chloride channel blocker), causing unopposed neuroexcitation and resulting in refractory seizures (25). Adenosine, a third critical neurotransmitter, is released by the presynaptic neuron during seizure activity and autoregulates further ictal discharge. Methylxanthines such as theophylline and caffeine, a once commonly prescribed asthma medication, inhibit endogenous adenosine and can lead to status epilepticus in overdose patients. While consideration of the crucial balance between neuroexcitation and neuroinhibition provides a framework with which to consider the poisoned seizure patient, one must keep in mind that any alteration of neuronal homeostasis can result in seizure. As such, toxins that lead to hypoglycemia, hypoxia, electrolyte abnormalities, or any other metabolic derangement may also cause seizure. Thus, the list of drugs that has been reported to cause seizure is too long to provide much clinical utility. Instead, the subsequent paragraphs highlight a select few toxins that characteristically cause consequential seizure activity in overdose or exposure.
Camphor, an essential oil once commonly found in mothballs, is a natural extract now used in nasal decongestant rubs and insect repellants. Children exposed to camphor often exude its unique aromatic odor. They may become symptomatic following ingestion or inhalation as well as through intranasal or transdermal absorption. Seizure is often the first manifestation of toxicity and occurs from minutes to hours after ingestion. It is typically short and self-limited although multiple seizures have been reported (26–29). The exact mechanism of camphor toxicity remains unclear but this lipophilic cyclic terpene may cause other neurologic manifestations such as CNS depression, agitation, headache, and hyperreflexia. The management strategy remains supportive, including administration of standard seizure medications starting with benzodiazepines.
Isoniazid, a commonly prescribed antituberculous medication, can cause refractory seizures in overdose and should be considered in any case of status epilepticus. It inhibits the enzyme pyridoxine phosphokinase, which is needed to activate pyridoxine to pyridoxal phosphate. Without this activated pyridoxine, GABA cannot be generated from glutamic acid, leading to a lack of physiologic inhibitory tone and status epilepticus. The treatment for this poisoning is IV pyridoxine (vitamin B6), of which 1 gram should be administered for every 1 gram of isoniazid ingested. If the amount of ingested isoniazid is unknown or if the antidote is administered empirically for status epilepticus, a dose of 70 mg/kg IV (maximum of 5 g) should be administered at 0.5 g/min until the seizure stops and the remainder given IV over 4 to 6 hours. Benzodiazepines should also be administered as they work synergistically with pyridoxine to increase GABA agonism. Hydrazines, chemicals found in Gyromitra mushrooms (“false morel”) or in rocket fuel, are structurally similar to isoniazid and can cause seizures by comparable mechanisms, also requiring treatment with pyridoxine (30).
While many commonly prescribed antidepressants have been reported to cause seizure in overdose, two drugs in this class deserve special attention. Citalopram, a selective serotonin reuptake inhibitor (SSRI), frequently causes seizure early after overdose. It may also cause QT prolongation on the ECG, which may be delayed up to 33 hours following ingestion (31,32). These effects appear to be dose-related and become more likely with larger overdoses. Bupropion, an atypical antidepressant often prescribed for smoking cessation, inhibits reuptake of norepinephrine and dopamine. It is associated with seizures at both therapeutic doses and in overdose, with an increased risk of delayed seizures and status epilepticus following ingestions of sustained release preparations. These patients are managed supportively with at least 24 hours of observation due to a consequential risk of delayed seizure following overdose.
Toxicologists are occasionally called to the pediatric intensive care unit to consult on previously healthy, school-aged children with abrupt onset of intractable seizures, typically occurring after a mild febrile prodrome, as described in a cohort of such children by Mikaeloff and colleagues (33). In addition to the induced pyridoxine deficiency-related intoxications noted above, a few additional relatively exotic toxic compounds are also associated with prolonged seizures that may be refractory to routine anticonvulsant therapy (34). These include the metals lead (e.g., lead encephalopathy, often associated with cerebral edema, and anemia with basophilic stippling) (35) and arsenic (especially in the context of acute arsenic poisoning, typically associated with severe gastroenteritis; cardiotoxicity with shock and /or QTc prolongation and torsades de pointes; and acute lung, liver, and renal injury) (36), methylxanthine medications such as caffeine and theophylline, and the biologic toxins cicutoxin (from the water hemlock plant), and domoic acid (in amnestic shellfish poisoning) (4). Thus, such patients warrant a careful medication, environmental, dietary, and parental occupational history and complete physical examination, with emphasis on seeking evidence of multiorgan system dysfunction, and notation of unusual physical findings such as alopecia and abnormal fingernails (e.g., Mees lines, suggestive of heavy metal exposure). Comprehensive laboratory analysis (e.g., complete blood count, comprehensive metabolic panel with renal and hepatic function tests, urinalysis, and ECG, as well as specific toxicological testing) should be considered, perhaps best effected in conjunction with toxicology or poison center consultation. Unfortunately, in the authors“ experience, such an evaluation, though reassuring in eliminating several potentially treatable conditions, has been unrevealing in most such patients.
Serotonin Syndrome
The serotonin syndrome, or serotonin toxicity is a constellation of signs and symptoms that result from excess stimulation of the serotonin type 1A receptors. This excess serotonergic agonism often results from the additive effects of drugs that enhance serotonin modulation. Some such drugs include those that inhibit serotonin metabolism (i.e., MAOIs), those that prevent reuptake of serotonin (i.e., SSRIs,) those that enhance serotonin release (i.e., MDMA), and those that directly act as agonists at serotonin receptors (i.e., lithium). Affected patients present with some combination of salient features including altered mental status, tremor, hyperreflexia of the lower extremities, muscle rigidity, diaphoresis, diarrhea, shivering, autonomic instability, and hyperthermia (37). The serotonin syndrome is a diagnosis made purely on clinical grounds, is one of exclusion, and often manifests on a spectrum with varying degrees of severity. The management of these patients mirrors that of the sympathomimetic patient, with special attention paid to actively cooling hyperthermic patients and providing benzodiazepines for agitation, tachycardia, and hypertension. The commonly cited antidote for this syndrome, cyproheptadine, acts as a serotonin antagonist but is not considered a first line therapy. It has not demonstrated clear benefits in patients with the serotonin syndrome and may cause anticholinergic effects including hyperthermia. It might be considered in patients who have failed to respond to good supportive care and benzodiazepines.
Neuroleptic Malignant Syndrome
The neuroleptic malignant syndrome, another drug-related hyperthermic syndrome, differs from serotonin syndrome both by the implicated drugs and by the insidious nature of its onset. While serotonin syndrome typically manifests within minutes to hours of treatment with a serotonergic agonist, neuroleptic malignant syndrome tends to evolve over the course of days following exposure to an implicated drug. Neuroleptic malignant syndrome results from dopamine antagonism in the CNS most commonly secondary to antipsychotic therapy. Higher potency agents such as haloperidol are more commonly causative especially in the setting of rapidly increasing doses and depot forms of administration (i.e., haloperidol decanoate). Clinical manifestations, similar to the serotonin syndrome, include altered mental status, hyperthermia, autonomic instability, tremor, and myoclonus. Increased muscle tone, also a finding common to both syndromes, can be distinguished in cases of neuroleptic malignant syndrome by its “lead pipe” quality. Treatment requires active cooling in the setting of hyperthermia and muscle relaxation with benzodiazepine therapy. If the patient fails to respond to these initial interventions, bromocriptine, a centrally acting dopamine agonist, should be considered.
Salicylism
Salicylism results from a wide variety of salicylate derivatives including acetylsalicylic acid (aspirin), methyl salicylate (oil of wintergreen), or occasionally bismuth subsalicylate (Pepto-Bismol.) While classically noted for its relatively unique acid-base derangement, a primary respiratory alkalosis coupled with a primary metabolic acidosis, close observation of the salicylate poisoned patient reveals a much more complex toxidrome than that found on a blood gas. Affected patients are both tachypneic and hyperpneic leading to the appearance of increased work of breathing in the setting of a clear pulmonary exam. Additionally they may complain of tinnitus or decreased hearing and exhibit nausea and vomiting along with diaphoresis and hyperthermia. The primary goal of therapy should be to prevent salicylic acid from crossing the blood brain barrier and entering the CNS where it can lead to cerebral edema and death. Alkalinization of the patient’s serum with sodium bicarbonate accomplishes this, causing the salicylic acid to remain in an ionized form that cannot permeate the blood brain barrier. In turn, the patient’s urine is alkalinized, similarly preventing reabsorption of this ionized acid in the distal tubule and enhancing elimination. The patient’s spontaneous respiratory alkalosis should also be preserved if possible, with avoidance of sedation and intubation if practical. In cases where endotracheal intubation is clinically necessary, the ventilator should be set to maintain an appropriately large minute ventilation, maintaining the respiratory alkalosis.
The recommended treatment of symptomatic salicylate poisoned patients includes alkalinization of the blood and urine, starting with 1 to 2 mEq/kg IV push over 1 to 2 minutes and followed by an infusion of 3 ampules sodium bicarbonate in 1 L of D5W run at twice maintenance. The infusion should be adjusted to reach a goal urine pH of 7.5 to 8 and a serum pH of 7.45 to 7.55. Serum potassium should be repleted and serial salicylic acid concentrations obtained until the concentration clearly trends down toward normal. Hemodialysis should be initiated if the patient cannot tolerate bicarbonate therapy (i.e., fluid overload), shows signs of end organ toxicity (i.e., altered mental status), has renal failure, or has an acute salicylate concentration of greater than 100 mg/dL or a chronic salicylate concentration of greater than 60 mg/dL (38). Of course, general signs of clinical deterioration in the setting of alkalinization also warrant drug removal by hemodialysis.
Tricyclic Antidepressant (TCA) Toxicity
TCA poisoned patients present with altered mental status and can rapidly deteriorate with the development of seizures, coma, cardiovascular collapse, and death. The ECG remains the cornerstone of diagnostic and prognostic significance of these patients. TCAs cause direct myocardial toxicity by binding to rapidly inactivating sodium channels on the myocardium, inhibiting sodium influx and slowing the phase 0 depolarization of the myocyte (Figure 17.1). In patients with a limb lead QRS interval of 100 milliseconds or longer, 33% have seizures. When the limb lead QRS interval is greater than 160 milliseconds, 50% develop ventricular dysrhythmias (6). Given this prognostic potential, all TCA poisoned patients should have an immediate ECG. If the QRS interval is 100 milliseconds or greater, the patient should receive a sodium bicarbonate bolus of 1 to 2 mEq/kg IV push over 1 to 2 minutes. If the QRS narrows in response to this therapy, repeat boluses of sodium bicarbonate should be given or an infusion titrated to keep a narrow QRS interval while maintaining the serum pH between 7.45 and 7.55. Seizures should be treated with benzodiazepines at standard doses.
■ CONCLUSION
The poisoned patient shares much in common with other critically ill patients. Such patients require the same attention to airway support, breathing, circulation, and core body temperature. They require a sound clinical approach which is not overcome by an all-consuming preoccupation with reversing the toxin. As is commonly quipped by toxicologists, “Treat the patient, not the poison.” The vast majority of poisoned patients do not require a specific antidote to ensure an optimal outcome, but rather they require both toxin specific therapy and general supportive care. Once the ABCDEFs have been addressed, contact with a regional poison center or institutional medical toxicologist is advised. Such consultation might be especially advisable in several recurring scenarios, based on the authors“ experience: any child known to be poisoned who requires escalating supportive care, antidotal therapy or enhanced elimination interventions; an undiagnosed critical illness of acute onset, with significant alteration of mental status, especially if accompanied by multi-organ system dysfunction; protracted or recurrent neurologic dysfunction that is not diagnosed after the “usual suspects” have been evaluated for and excluded; and any particularly puzzling clinical picture, especially in toddlers and adolescents (or, if such occurs in young infants, or school-aged children, the possibility of a toxic etiology for Munchausen syndrome by proxy might be considered). The medical toxicology service in these situations may be able to narrow the differential diagnosis of the poisoned patient and can offer specific management strategies for particular poisonings.
■ REFERENCES
1. et al. Environmental factors associated with a spectrum of neurodevelopmental deficits. Ment Retard Dev Disabil Res Rev. 2002;8(3):188–197.
2. , . Neurodevelopmental toxicology. Neurol Clin. 2005;23(2):321–336.
3. . Neurologic concepts of lead poisoning in children. Pediatr Ann. 1992;21(6):384–388.
4. . Neurologic principles. In: Goldfrank’s Toxicological Emergencies. 9th ed. New York, NY: The McGraw Hill Companies; 2011:275.
5. . Antidiabetics and hypoglycemics. In: et al, eds. Goldfrank’s Toxicological Emergencies. 9th ed. New York, NY: McGraw Hill Companies; 2011:714.
6. , Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313(8):474–479.
7. , , . Severe hyponatremia and inappropriate antidiuretic hormone secretion following ecstasy use. Acad Emerg Med. 1998;5(8):839–840.
8. et al. Hyponatraemic states following 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) ingestion. QJM. 2002;95(7):431–437.
9. . A flood of opioids, a rising tide of deaths. N Engl J Med. 2010;363(21):1981–1985.
10. . Naloxone reversal of buprenorphine-induced respiratory depression. Clin Pharmacol Ther. 1989;45(1):66–71.
11. et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology. 2006;105(1):51–57.
12. , , . Naloxone reversal of opioid-induced respiratory depression with special emphasis on the partial agonist/antagonist buprenorphine. Adv Exp Med Biol. 2008;605:486–491.
13. . Flumazenil and seizures: analysis of 43 cases. Clin Ther. 1992;14(2):292–305.
14. et al. Excited delirium, restraints, and unexpected death: a review of pathogenesis. Am J Forensic Med Pathol. 2010;31(2):107–112.
15. , , . Positional asphyxia, a cause of death insufficiently known. Rev Med Suisse. 2011;7(303):1511–1514.
16. , . Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217(2):350–356.
17. et al. Ambient temperature and mortality from unintentional cocaine overdose. JAMA. 1998;279(22):1795–1800.
18. et al. Whole-body cooling of hyperthermic runners: comparison of two field therapies. Am J Emerg Med. 1996;14(4):355–358.
19. et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112(12):897–903.
20. et al. Datura stramonium L. poisoning in a geophagous child: a case report. Int J Emerg Med. 2011;4(1):31.
21. Centers for Disease Control and Prevention (CDC). Jimsonweed poisoning associated with a homemade stew—Maryland, 2008. MMWR Morb Mortal Wkly Rep. 2010;59(4):102–104.
22. et al. Jimson weed abuse in an Oklahoma teen. J Okla State Med Assoc. 2009;102(12):351–353.
23. , . Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9(11):588–590.
24. . Mushroom poisoning in infants and children: the Amanita pantherina/muscaria group. J Toxicol Clin Toxicol. 1992;30(1):13–22.
25. et al. Status epilepticus from an illegally imported Chinese rodenticide: “tetramine”. J Toxicol Clin Toxicol. 2003;41(7):991–994.
26. . Camphor ingestion: an unusual cause of seizure. J Assoc Physicians India. 2009;57:216; author reply 216–217.
27. , , . Camphor poisoning following ingestion of mothballs ‘for headache’. Ned Tijdschr Geneeskd. 2011;155(39):A3676.
28. et al. A cluster of children with seizures caused by camphor poisoning. Pediatrics. 2009;123(5):1269–1272.
29. , , . Prolonged seizures due to contact and inhalation exposure to camphor. A case report. Clin Pediatr (Phila). 1977;16(10):901–902.
30. , , . Pyridoxine in clinical toxicology: a review. Eur J Emerg Med. 2005;12(2):78–85.
31. et al. Prolonged QTc interval due to escitalopram overdose. J Miss State Med Assoc. 2010;51(12):350–353.
32. , , . Citalopram overdose: late presentation of torsades de pointes (TdP) with cardiac arrest. J Med Toxicol. 2008;4(2):101–105.
33. et al. Devastating epileptic encephalopathy of school-aged children (DESC): a pseudo-encephalitis. Epilepsy Res. 2006;69:67–79.
34. , . Refractory seizures. Pediatr Emerg Care. In press, 2012.
35. . Lead. In: et al, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill Companies; 2011:1266.
36. , . Arsenic. In: et al, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill Companies; 2011:1214.
37. , . The serotonin syndrome. N Engl J Med. 2005;352(11):1112–1120.
38. . Salicylates. In: et al, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill Companies; 2011:508.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


