Another subset during the first year of life includes those previously healthy infants who unexpectedly develop a precipitous neuromuscular crisis. Specific clinical mechanisms, that require consideration in this instance, include a previously unrecognized SMA, GBS, infantile botulism, congenital myasthenic syndromes, or, very rarely, postvaccine poliomyelitis.
Anterior Horn Cell
The Werdnig-Hoffmann form of SMA or SMA type I is one of the most common causes of the floppy infant syndrome as well as the most common anterior horn cell disorder of infancy (2,3). Usually, these infants either present as a floppy baby, or with failure to develop or loss of early motor milestones between 1 and 3 months of age. Rarely, SMA (classified as type 0) may present at birth with profound hypotonia, severe weakness, and respiratory failure (5,6). Eventually some SMA type I infants will later require neonatal ICU. Their diagnosis is usually made by a combination of EMG and DNA analysis or just DNA analysis (2). Occasionally, their SMA is not initially recognized and it is not until they are unable to cope with a respiratory illness that they present to the ICU requiring pulmonary support or cannot be weaned from a ventilator.
Case 1: Werdnig-Hoffman Disease Presenting With Respiratory Distress
This previously “healthy” 4-month-old first child was admitted with an acute bronchiolitis and required intubation. Subsequently, when his pulmonary status had significantly improved his pediatricians had unexpected difficulty weaning him from the respirator. Neurologic consultation demonstrated generalized hypotonia and absent muscle stretch reflexes.
An EMG was performed because of its ability to provide a rapid means to localize the anatomic site for the baby’s motor unit abnormality. This demonstrated low amplitude compound muscle action potentials (CMAPs), normal motor conduction velocities (MCVs), and distal latencies (DLs), as well as sensory nerve action potentials (SNAPs). Needle EMG demonstrated a marked decrease in the number of motor unit potentials (MUPs) with widespread active denervation characterized by many fibrillation potentials and positive waves. Subsequent to the EMG, DNA analysis for the SMA defect at 5q11-13 was positive (homozygous deletion of exon 7 of the survival of motor neuron 1 [SMN1] gene, which is the most common mutation in SMA patients).
Comment
Infants with SMA are usually easily recognized because of their typical clinical presentation as a floppy baby with an alert facies, absent muscle stretch reflexes, and fasciculations of their tongue. In this instance, DNA analysis of the SMN1 gene is the appropriate means to make the diagnosis today. Occasionally, however, parents and pediatricians have not recognized a relatively subtle clinical presentation until an acute illness leads to hospitalization. In the intensive care setting, as above, one may need to make a more rapid diagnosis, utilizing EMG, to provide appropriate clinical management as well as counseling of the parents. Rarely, infants with non-5q SMAs may have similar clinical and EMG findings (7).
Another form of acute anterior horn cell disease, vaccine-associated poliomyelitis, deserves mention in the differential diagnosis of any infant or child with an acute weakness (8).
Case 2: Acute Postimmunization Poliomyelitis
A previously healthy 11-week-old boy presented with a 2-day history of fever, irritability, lethargy, and head lag. Computerized tomographic (CT) examination of the head and brain was normal. CSF analysis demonstrated 580 white blood cells (WBCs), 41% polymorphonuclear leukocytes, 59% lymphocytes; glucose 59 mg/dL, and protein 143 mg/dL. Although intravenous antibiotics were initiated, the baby soon developed apneic spells requiring intubation. This infant soon developed a weak cry and asymmetric skeletal muscle weakness particularly sparing left plantar flexor and extraocular muscles.
On the seventh day of his illness an EMG was performed demonstrating an anterior horn cell disease pattern. Subsequently, repetitive motor nerve stimulation (RMNS) failed to identify a neuromuscular transmission defect (NMTD).
Review of this baby’s immunization records documented that he had received type 3 Sabin live poliomyelitis vaccine 3 weeks prior to onset of his illness. His recovery was poor; he was ventilator dependent more than 2 years later.
Comment
Three cases of acute flaccid paralysis occurring in infants, secondary to immunization-related poliomyelitis, became known to one of the authors (HRJ) within just a few years during the mid-1990s (8,9; J. Goldstein, personal oral and written communication, 1995). All three babies were 3 to 4 months old, each having received type 3 polio immunization less than 1 month earlier. An acute febrile illness preceded a progressive asymmetric extremity weakness, head lag, irritability, and lethargy. A CSF pleocytosis with 100 to 580 WBCs was associated with protein values between 82 and 143 mg/dL in all and glucose of 49 mg/dL in 1 (8). EMG demonstrated classic electrophysiologic evidence of an acute asymmetric anterior horn cell disorder.
Most instances of paralytic poliomyelitis from the attenuated live Sabin oral vaccine occurred among infants less than age 1 year usually subsequent to their first polio vaccine, and within 2 weeks of immunization (10). Revised Center for Disease Control guidelines require that the initial immunization protocol utilize Salk’s killed vaccine as the primary immunization method for all children (11). This new infantile immunization protocol has made postvaccination related poliomyelitis of historical interest in North America. However, the live vaccine is still being utilized in underdeveloped countries.
Peripheral Nerve
Some congenital hypomyelinating or axonal polyneuropathies present in the newborn period with a clinical phenotypic appearance similar to SMA I, Werdnig-Hoffman disease. These infants may be so ill that they require intensivist support (2).
Case 3: Severe Newborn Infantile Demyelinating Polyneuropathy
A 2-week-old old infant was delivered after what the mother perceived as a normal pregnancy with normal fetal movements in utero. Since birth, he had a “paucity of spontaneous movements,” and a weak cry. His mom stated that he was still able to breast-feed although he did not hold the pacifier tightly. His gag was weak. Fasciculations of the tongue were present. This alert baby was markedly hypotonic with frog leg posture, no spontaneous movements of the legs, wrist drops, weak palmar grasp, and arthrogryposis multiplex. He assumed the inverted U posture when held prone. He was areflexic and the plantar responses were mute. He responded to noxious stimuli applied to all extremities.
The CSF protein was elevated at 190 mg/dL. Sensory NCS failed to demonstrate any median SNAP. Median and peroneal CMAPs were widely dispersed, of very low amplitude 0.05 and 0.02 mV, had prolonged latencies 3.6 and 8.2 ms and very slow MCV of 6 and 8 m/sec. EMG demonstrated mild diminution in the number of MUPs which were of slightly longer duration, polyphasic, normal amplitude, and firing at increased frequency. The only areas with occasional fibrillations and positive waves were present in the most distal muscles. These findings were compatible with an acquired, diffuse, predominantly demyelinating, peripheral neuropathy.
Sural nerve biopsy demonstrated hypomyelination with abortive onion bulb formation. These findings did not provide a base to distinguish between an inborn error of myelination or an unusual acquired autoimmune demyelinating peripheral neuropathy, such as intrauterine GBS. A trial of prednisone was unsuccessful. Subsequently, progressive respiratory failure ensued. The baby died at age 2 months; an autopsy was declined.
Comment
Any floppy infant presenting with a frog leg posture and tongue fasciculations most likely has SMA type 0 or type I. In contradistinction, this baby’s EMG findings of very slow MCVs, with dispersed low amplitude CMAPs, and absent SNAPs were diagnostic of an acquired demyelinating polyneuropathy and not an abnormality primarily at the motor neuron level. The incidence of peripheral neuropathies was 6% in our BCH review of over 100 floppy babies seen in our EMG lab (2,3). These were more common among the infants who also had arthrogryposis (3,12). The absence of sensory responses is the most important pediatric EMG clue to the appropriate diagnostic conclusion that the baby has one of these rare neonatal neuropathies. The nerve pathologies included primary axonal, demyelinating, and widespread neuronal degeneration.
GBS rarely affects newborn infants. It needs consideration in the differential diagnosis of neonatal flaccid paralysis (13–15). In one instance, GBS onset was soon after birth (13). The baby’s mother had an autoimmune disease, chronic ulcerative colitis. During the 30th week of her pregnancy she had observed significantly decreased fetal movements (13). Her baby was quadriparetic when delivered at 37 weeks with an Apgar score of 6 at 10 minutes. At age 3 days, neurologic exam demonstrated severe leg and moderate proximal arm weakness, with generalized absence of muscle stretch reflexes. The CSF protein was normal, 38 mg/dL. In contrast, motor NCS demonstrated profound motor nerve conduction velocity (NCV) slowing between 3 and 15 m/sec, with conduction block and temporal dispersion in many nerves. Needle EMG revealed active denervation in many muscles. He had gradual and complete recovery by age 1 year without any specific treatment (13).
Another pregnant mother, aged 33, developed severe GBS during the 29th week of her pregnancy (14). She became tetraplegic and was still respirator dependent when her baby was born at the 38th week. He developed hypotonia, marked respiratory distress, and feeding problems 12 days postpartum although he was well at birth. His CSF protein was 243 mg/dL. An EMG was typical for GBS. Both the mother and child had immunoglobulin G (IgG) antibodies positive for recent cytomegalovirus infection. Intravenous immunoglobulin therapy was associated with a complete symptomatic resolution in 2 weeks (14). One presumes his GBS developed in utero (14).
GBS affected another pregnant woman. Her baby had poor spontaneous ventilation at delivery requiring brief intubation. During the next 5 days of observation, there were no other signs of GBS (15). Despite such, both this infant and his mother had high antibody titers to human peripheral nerve myelin glycolipids (15).
Even without any known intrauterine events, GBS may occur shortly after birth, as early as age 3 weeks. These previously healthy babies present with an acute, rapidly progressive, and often severe hypotonia, with possible respiratory distress, and feeding difficulties (16,17). Chronic inflammatory demyelinating polyneuropathy subsequently developed in a 7-week-old infant with acute GBS (18). An EMG is particularly helpful for the differentiation of GBS from other acute floppy infant syndromes (16). Absent SNAPs, profound motor conduction slowing, and dispersed CMAPs are the quintessential EMG clues for the diagnosis of neonatal GBS. In our BCH EMG review of more than 100 floppy babies, there was a 6% incidence of peripheral neuropathies (3). Only one of these six infants had EMG findings commensurate with GBS (3).
Although tick paralysis mimics acute GBS in children, we are unaware of an infantile case. Nonetheless, any baby with an acute onset of generalized muscle weakness needs to be searched for a tick, particularly in the scalp (19).
Neuromuscular Junction
Various neuromuscular junction (NMJ) disorders are occasionally encountered during infancy. Autoimmune maternal associated MG is the most easily recognized and probably most common NMJ pathologic entity occurring in this age group. This relatively short-lived disorder typically occurs at birth in 15% to 20% of infants whose mothers have MG (20–22). Rarely, infants born to mothers who had eclampsia and received magnesium treatment, also have the potential to develop an acute infantile presynaptic NMJ disorder (23,24). These two neuromuscular transmission disorders only occasionally come to the attention of the pediatric neurologist as pediatricians are well versed in the management of these infants.
There are also a number of relatively rare congenital myasthenic syndromes with a primary pathophysiologic site of difficulty varying between the presynaptic NMJ, the synapse, or the postsynaptic NMJ. An endplate deficiency of choline acetyltransferase is a congenital myasthenic syndrome that predisposes the baby to an acute onset NMJ disorder (25). Typically, these babies have recurrent episodic apnea. In contrast, it is unusual for other congenital myasthenic syndrome forms to have such an acute presentation.
Infantile botulism is the one other presynaptic neuromuscular transmission disorder that will acutely affect previously healthy babies and toddlers. Clinical suspicion, with subsequent EMG, is crucial to making this diagnosis. Because of its somewhat varied presentation we are concerned that possibly infantile botulism is more common than recognized.
Case 4: Infantile Botulism
A previously healthy, 4-month-old baby was admitted to another hospital with a 1-week history of constipation, and subsequently poor feeding, lethargy, a weak cry, and dehydration. She had lost her ability to roll over, lift her head, or sit up. A full septic evaluation was unremarkable including CSF examination. After she developed cyanosis with an acute apneic spell, and subsequent increasing lethargy, she was transported to BCH.
Attempt to wean her from the ventilator was unsuccessful. Neurologic examination here demonstrated no extraocular muscle movements, absent corneal responses, and no gag reflex. At this time she still had movement in all extremities, and intact muscle stretch reflexes. The initial clinical impression was an “indeterminate encephalopathy.” This conclusion was based on nonspecific EEG findings in the right occipital lobe, as her brain magnetic resonance imaging (MRI), CSF, bacterial and viral cultures, and metabolic evaluations were all normal. During this evaluation, she became increasingly hypotonic and areflexic. Seven days after onset of symptoms and subsequent to further clinical review, at change of service a diligent intern requested an EMG for possible infantile botulism.
The EMG evaluation, in the ICU, demonstrated low amplitude median and peroneal CMAPs (0.56 mV and 0.2 mV respectively; normal > 3.5 mV and 1.6 mV) with normal motor NCV, and DLs. RMNS of the median nerve at 2 Hz demonstrated an 11% decrement, and with 20 Hz stimulation a 160% facilitation. MUPs were very “myopathic.” These EMG findings were consistent with a diagnosis of infantile botulism. Subsequently, a stool sample, as well as an assay of a honey specimen from home, were both positive for type A botulinum toxin. She gradually improved, with supportive treatment, during the next few months.
Comment
Infantile botulism usually has a stereotyped clinical presentation. Typically, a previously healthy infant, between ages 10 days and 12 months, has the acute onset of hypotonia, generalized descending weakness, poor feeding, oculomotor and pupillary abnormalities, dysphagia, and constipation (26–33). However, one also needs to consider this diagnosis in the setting of unexplained respiratory distress in any baby under 12 months of age. Infantile botulism has been linked to honey ingestion. In its most severe state the presentation may be acute and require ventilatory support. Treatment with human botulinum antitoxin can expedite the infant’s recovery (34,35).
Bacteriologic confirmation usually requires a few weeks, but a mouse bioassay and real-time polymerase chain reaction (PCR) assays can provide a rapid confirmation of the diagnosis (36). EMG is the most useful early diagnostic tool. Most babies with infantile botulism have clear-cut evidence of a neuromuscular transmission disorder. Low-rate RMNS, at 2 to 5 Hz, often but not always demonstrates a decremental response (37). More rapid RMNS rates of 20 to 50 Hz provide the primary electrophysiologic technique for diagnosing infantile botulism (37). A posttetanic facilitation varying between 23% and 313%, with a mean of 73%, was documented in 23 of 25 babies (37). A longer period of posttetanic facilitation is also observed. One may not always be able to confirm a diagnosis of infantile botulism with EMG as not all cases have a documented facilitation with RMNS (37,38). However, in most instances of infantile botulism, babies will have a typical EMG diagnostic triad (39). This includes: (1) low amplitude CMAPs (beware normal maturation standards for age), (2) posttetanic facilitation, and (3) absence of posttetanic exhaustion (38).
Muscle
Congenital myopathies typically present as a floppy infant syndrome but most babies are not ill enough to require neonatal ICU monitoring. In addition, myopathies do not typically cause acute infantile flaccid paralysis and/or respiratory distress. However, occasionally we are asked to see a neonatal ICU baby who is found to have a primary myopathy.
Case 5: Neonatal MD
Respiratory distress developed soon after birth in this baby girl, with clubfeet. She required intubation for her first month of life. An EMG was requested for evaluation of her generalized hypotonia when she was 2 months old. Motor NCS demonstrated low amplitude CMAPs but other parameters were normal including SNAPs. Profound and prolonged decrescendo myotonic discharges occurred during her EMG. These were associated with rare fibrillation potentials. Her MUPs were normal. The mother had no myotonia detectable by physical examination or on EMG but she recalled that her father had difficulty releasing his grip when driving a car.
Comment
MD is one of the few dystrophies that presents as a severely floppy infant. Babies with congenital MD may have severe generalized hypotonia requiring intubation at birth for their associated respiratory compromise. Sometimes, the diagnosis is suspected clinically by their classic facies with “tenting” of the mouth that appears like an upside down letter V. Most all MD infants have a family history of MD, however it is often unrecognized in the parent. Typically, the mother has a milder form requiring premature cataract removal or has evidence of mild subclinical myotonia and/or distal weakness on clinical exam (40).
Today when confronted with the typical clinical phenotype, DNA testing is 98% to 99% accurate in both infants with MD as well as their symptomatic or asymptomatic mothers. Myotonia detectable by EMG may be present in the hypotonic newborn infant, as early as age 5 days, although it sometimes is not always evident this early on (41,42). Occasionally, one finds that a maternal EMG will detect previously unsuspected myotonia (2). Nowadays, a DNA test will quickly establish the diagnosis.
Case 6: Metabolic Myopathy: Phosphofructokinase Deficiency
A 39-week gestation newborn infant had a history of diminished fetal movement and developed immediate respiratory distress at birth. He had severe generalized and bifacial weakness, a high arched palate, absent muscle stretch reflexes, and marked distal arthrogryposis. At 2 days of age, NCS demonstrated low amplitude to absent CMAPs with otherwise normal motor and sensory conduction parameters. EMG demonstrated very low amplitude, short duration MUPs primarily in proximal muscles typical of a myopathy. Muscle biopsy demonstrated an absence of the glycolytic enzyme phosphofructokinase. Swoboda and colleagues (43) suggested that a ketogenic diet might be beneficial. This commenced at age 4 months. He gained some clinical motor improvement through age 2 years; however, he died of other complications later that year.
Comment
Metabolic myopathies rarely present with severe hypotonia and respiratory weakness in the newborn period. A fatal case of phosphorylase deficiency presented at 4 weeks of age with feeding difficulties, tiring easily, and severe respiratory distress. This infant had a rapidly progressive course, becoming very hypotonic with generalized weakness (44). A muscle biopsy shows an excess of glycogen accumulation in infants with phosphofructokinase deficiency; the diagnosis is confirmed with enzyme assay and/or genetic analysis.
■ ACUTE NEUROMUSCULAR CRISES IN THE TODDLER AND THE OLDER CHILD
Anterior Horn Cell
We are pleased to comment that since the inception of our EMG lab in 1979 at BCH we are not aware of the occurrence of any acute poliomyelitis, beyond infancy. However, one needs to keep this possibility always in mind, as there are some parents who reportedly still decline to have their children appropriately immunized. In addition, with the increased use of air travel and children being adopted, immigrating, or visiting from all parts of the world, one always needs to be alert to the possibility of poliomyelitis in countries with less than ideal immunization standards. However, we do see a few instances of subacute polio in children coming from those nations where standard immunization is not routinely available (45). Physicians must keep poliomyelitis within the differential diagnosis of any child with an acute, particularly asymmetric, flaccid paralysis.
Peripheral Nerve
Today, GBS is the most common cause for an acute generalized paralysis in the older child. A few rare other primary pediatric polyneuropathies occasionally develop. In addition, some of these clinical settings are caused by some unusual neuromuscular transmission defects as noted later in this chapter. In the ICU setting, an EMG is particularly important for rapid localization of the acute motor unit lesion. It may also prove helpful for making prognostic judgments. The following case examples illustrate some of the interesting GBS variants.
Case 7: Guillain-Barré Syndrome: Pseudoencephalopathy, Painful Weakness, and Meningismus
Two weeks prior to evaluation at BCH, this previously healthy 5-year-old boy participated in an Audubon field trip to collect insects along the Rhode Island coast. He soon developed a fine papular rash on his face and neck that was followed 24 hours later by headache, nausea, and vomiting during a family sailing cruise. Bilateral pain developed in his knees, posterior thighs, and popliteal fossa 4 days prior to our evaluation. A viral syndrome was diagnosed and the pediatrician treated him symptomatically. Subsequently he lay in his bunk complaining of headaches and occasional vomiting but no fever. He was brought to BCH 1 week later.
Here he was extremely uncomfortable, groaning and writhing. His conjunctivae were injected, and his neck stiff. Muscle strength was “grossly” normal, and capable of supporting his weight to stand. Sensation seemed intact, however, his muscle stretch reflexes were not elicitable. Brain CT was normal. Subsequent CSF analysis demonstrated a protein of 145 mg/dL, glucose 65 mg/dL, and 1 WBC/dL. On neurologic examination the next morning he was listless with bilateral Babinski signs. Muscle strength was still difficult to assess.
Motor NCSs were of variably low amplitude with dispersed CMAPs, and multifocal asymmetric prolongation of DLs. Motor NCV varied from 22 m/sec for median to 38 m/sec for ulnar and 50 m/sec for peroneal fibers. Peroneal F waves were absent. No median or ulnar SNAPs were elicited. Needle EMG was normal. This EMG was compatible with an acute demyelinating polyneuropathy typical for GBS.
Comment
With the history of headache and rash, in an agitated child with Babinski signs, an encephalopathic process was the initial diagnostic consideration. After the brain CT was normal, a spinal fluid examination was performed. The high CSF protein led to the consideration of an atypical form of GBS. Other neurologic considerations included a transverse myelitis, an entity that also occasionally mimics GBS, however, there was no evidence of a cord level in this instance. Spinal MRI was therefore not performed. Some individuals with GBS appear to occasionally have an associated, but indeterminate central nervous system process such as locked-in syndrome, indicated by the presence of acute tetraplegia, areflexia, and cranial nerve involvement despite otherwise typical clinical and EMG findings for GBS. These patients, however, are fully conscious (46).
Severe pain was the primary presenting symptom of GBS in 21% (5 of 24) of children seen at BCH (47). A significant degree of pain was present in 67% of our GBS cases (47). At times, as in this instance, this discomfort was so bothersome and led to such irritability, that a number of these children were initially thought to have a meningoencephalopathy (48). Because of the severity of the pain, three of the children, each younger than this boy, were unable to participate in formal muscle testing. Thus the actual muscle weakness related to their GBS was difficult to clinically appreciate. A total of nine youngsters seen here at BCH were thought to be encephalopathic during their initial emergency room evaluations but later proven to have GBS (48). Initially each one received either brain CT or MRI, and when normal, this was followed by CSF exam anticipating an infection. When the albuminocytologic dissociation was found, neurologic consultation followed. The elevated CSF protein with areflexia and severe pain provided the clinical clues suggesting GBS. EMG was requested for further definition.
A reduced CMAP, in 83%, with absent or prolonged long latency waves, in 81%, were the two most common EMG findings in children with the demyelinating form of GBS (also known as acute inflammatory demyelinating polyneuropathy [AIDP]) seen at BCH (47). Specific findings consistent with demyelination, including conduction block, temporal dispersion, or MCV slowing, occurred in 70% to 74% of these children. MRI of the spinal cord may show enhancement of the nerve roots. In addition to GBS, the differential diagnosis of an acute demyelinating peripheral neuropathy in children includes some toxins, in particular glue sniffing, and rarely buckthorn wild cherry ingestion, insecticides, and thallium (16, 49). Porphyria, arsenic, lead, or mercury poisoning also enter the differential diagnosis when there is concomitant gastrointestinal distress (16). The differential diagnosis of children living in agricultural communities also includes organophosphate pesticide poisoning (50). Interestingly, in Paraguay a 30% incidence of organophosphate pesticide exposure was noted with otherwise typical childhood GBS (51). A few inborn metabolic errors, porphyria or Leigh disease (52), enhanced by medications, particularly barbiturates, may also precipitate symptoms resembling childhood GBS.
The clinical course and eventual prognosis of either the demyelinating or axonal forms is determined by the extent of primary or secondary axonal damage. There is a primary axonal variant of GBS that has been predominantly reported from China (53,54) but also occurs in America (particularly in Latin America), Europe, and Japan. In this instance, there is an initial immunologic reaction against epitopes specific to the axonal membrane. Axonal GBS has been classified into two subtypes: acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN) (55). EMG and NCSs are the main method for distinguishing AMAN, AMSAN, and AIDP; however, in certain cases, the distinction between AMSAN and AIDP may be difficult. Both AMAN and AMSAN may follow Campylobacter jejuni enteritis. Anti-GM1, anti-GM1b, and anti-GD1a IgG antibodies can be detected in patients with AMAN and AMSAN and can be used to differentiate these axonal forms of GBS from AIDP (56,57).
Case 8: Axonal GBS With “Brain Death” Presentation
A 6-year-old boy presented with a 2-week history of headaches and 2 days with difficulty walking. He had surgery and chemotherapy for a medulloblastoma 8 months previously, and subsequently was feeling fine. His last vincristine, cisplatin, and cyclophosphamide were completed 2 months earlier. Suddenly, he developed acute gasping and shortly thereafter had a respiratory arrest. On admission to the hospital he had no spontaneous movements and he was unresponsive to all noxious stimuli. He had total external ophthalmoplegia and his pupils were fixed and dilated. No muscle stretch reflexes or corneal, oculocephalic, and gag responses were elicitable. Plantar stimulation was mute. Initially he was thought to be “brain dead.” His CSF had a protein 167 mg/dL with just 6 WBCs. When an EEG was unexpectedly normal, a toxic screen and testing for heavy metals and porphyrins were also unremarkable.
An EMG was first performed on the fifth day of his illness. No motor or sensory responses, or MUPs were identified. However, fibrillations and positive waves were present in all muscles. Subsequent trial of intravenous immunoglobulin therapy did not result in any improvement for a few weeks. Eventually his cranial nerve function slowly recovered followed by respiratory and limb movements. One year later he was able to ambulate with a walker. His arm strength was then normal.
Comment
It is very rare to see fixed dilated pupils with GBS as occurred here. Both diphtheria and botulism are diagnostic considerations with this clinical set particularly when there is predominant bulbar involvement (58). MG or poliomyelitis also enters into this differential diagnosis but in these instances the pupillary responses are preserved.
Rarely, infectious diseases also need consideration in the differential diagnosis of GBS. In adults, GBS may be the presenting manifestation of AIDS. A 6-year-old boy with congenital AIDS has also developed GBS (59). However, to date based on a complete National Library of Medicine computer search, GBS has not been the presenting illness leading to a diagnosis of childhood AIDS. Lyme disease may be associated with a painful neuropathy, but this review also did not uncover an instance of a GBS-like illness in children. Diphtheria still occurs in those geopolitical settings without access to modern immunizations. Bulbar symptoms usually predominate early on. Usually these children have had a recent severe sore throat, and fever before the onset of the bulbar palsy. The acute peripheral neuropathy symptoms always occur subsequent to the onset of the bulbar symptoms. Once again as with the rare instance of infantile polio, the investigation of an immunization history is of significant importance in this clinical setting. Rarely one may see this constellation of findings in an older child. Diphtheria occurred in a teenager, presenting with bulbar symptoms, who was seen by one of us (HRJ). He had inadvertently received only one diphtheria, pertussis, and tetanus (DPT) immunization early in life (Table 21.2).
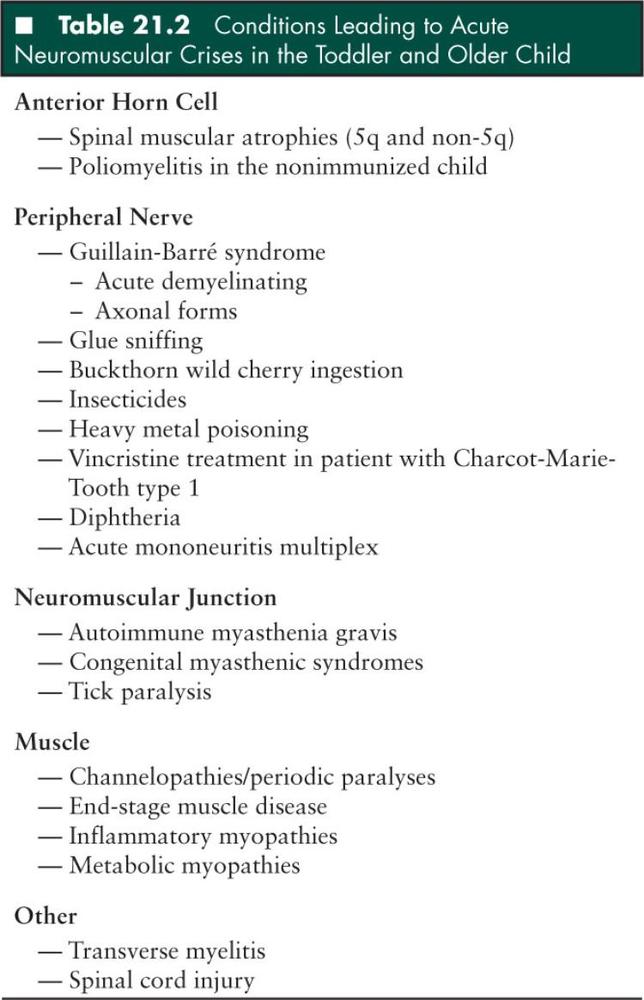
An acute vincristine exacerbation of an underlying polyneuropathy may occur if this chemotherapeutic agent is prescribed to children with a mild, subtle, or even unrecognized Charcot-Marie-Tooth type 1 (CMT1) hereditary neuropathy. This toxic neuropathy may be relatively severe when superimposed on this underlying genetic neuropathic process. As sometimes CMT1 is unsuspected early in life, it is important to query the parents before any vincristine therapy is initiated, as to the presence of any personal or family history of CMT1 by asking about the presence of high arches and weak feet. Testing for the specific genetic defect (a 17p11.2-12 duplication of the PMP22 gene) needs consideration in any child where there is any clinical suspicion of CMT1 prior to treatment with vincristine. One clinical example of such a severe neuropathy occurred in an immunocompromised child with an unrecognized family history of CMT1 (60).
Additionally, as mentioned earlier, children very occasionally present with one of two very rare, acute axonal forms of GBS. One of these, acute motor axonal neuropathy (AMAN), occurs predominantly in China (53), although it does have a much less common worldwide distribution. We have seen a few instances of this at BCH. Additionally, as discussed earlier, there is also an even less common acute motor sensory axonal neuropathy (AMSAN), which we have not diagnosed at BCH thus far. Another very rare pediatric form of GBS, originally defined by Miller-Fisher, also occurs in children (61). These children typically present primarily with gait ataxia, extraocular muscle palsies, and areflexia. Such a presentation occurred in about 5% of our BCH experience (47). Overlap cases, in which the clinical features of GBS and Miller-Fisher syndrome are intermixed, do occur. Miller-Fisher syndrome is also a postinfectious syndrome, frequently triggered by C jejuni enteritis. Sera from more than 90% of pure Miller-Fisher syndrome and GBS overlap patients, collected during the acute phase of the illness, contain high titers of antibodies to GQ1b ganglioside (62–65). These antibodies tend to disappear during clinical recovery. Strains of C jejuni isolated from Miller-Fisher syndrome patients bear GQ1b epitopes, suggesting that the underlying mechanism is one of molecular mimicry (66).
Prognosis in these pediatric GBS variants is determined by a combination of clinical and/or EMG signs of very significant axonal damage. Often children with GBS have a relatively benign prognosis (47). In some clinical instances, however, a more prolonged recovery, or even rarely a fatality (67), may occur. Examples include the child who requires a respirator, those in whom one cannot evoke a CMAP with NCS during an EMG, and those children who have very widespread active denervation on needle EMG. This last group may often require more than a year for reinnervation and subsequent recovery (58).
The prognosis for recovery in children with GBS is far better than for adults; however, there is no universal agreement on this opinion (68). Approximately 15% of hospitalized children with GBS require mechanical ventilatory support during the course of their admission (69). It is important to emphasize the need for the intensive-care setting, where it is possible to monitor cardiovascular and autonomic function (70). Of nine patients who died of GBS, two were children, one of whom died of cardiac arrest; the other had periods of bradycardia and hypotension and became neurologically unresponsive (67). Fluctuating blood pressure and autonomic instability have been recognized as predictors of a potentially fatal cardiac arrhythmia (67). Careful monitoring of respiratory function, good management of fluid balance, and avoidance of bed sores and pressure neuropathies are other important areas for care of the child with GBS. A significant cause of death in adults is pulmonary embolism; however, the literature is not clear as to the value of subcutaneous heparin for the prophylaxis of venous thrombosis in a child (16).
Guidelines as to when to transfer a child with GBS to the ICU and when to proceed with intubation have not been developed. Respiratory failure should be expected in any GBS patient with progressive appendicular or bulbar weakness. In children with GBS cared for on a regular pediatric ward, pulmonary insufficiency may develop insidiously and remain clinically silent without obvious signs of respiratory failure. Hypoxemia may not be detected until the vital capacity has fallen significantly, and hypercarbia is a late finding; therefore, blood gases are not useful in monitoring the diaphragmatic weakness in GBS patients. In older children who can cooperate for pulmonary function testing, bedside measurements of vital capacity (VC) and maximal inspiratory (PImax) and expiratory (PEmax) pressures should be carried out every 6 to 8 hours. In the younger uncooperative child, oxygen saturation, especially in sleep, should be monitored by pulse oximetry. It should be noted, however, that a fall in oxygen saturation is a late finding in children receiving oxygen.
Again, practice parameters allowing the prediction of need for mechanical ventilation have not been established in children. However, even in the adult literature the respective guidelines are not unanimous. A recent study summarizing 20 years of ICU experience from the Mayo Clinic, Rochester, Minnesota, provided some important guidelines which could be extrapolated to the pediatric age group (Table 21.3). Important clinical indicators of impending respiratory failure included bulbar dysfunction and autonomic instability, bilateral facial palsy, and rapid disease progression. The presence of these clinical parameters was associated with increased likelihood of mechanical ventilation. In our experience, however, bilateral facial palsy in children has not been a strong predictor of impending respiratory failure. Analysis of serial respiratory function measurements showed that a 30% reduction in VC, PImax, or PEmax were associated with subsequent progression to respiratory failure and intubation. Also, the following “threshold” respiratory values—VC less than 20 mL/kg, PImax less than 30 cm of H2O, and PEmax less than 40 cm of H2O—were shown to be highly associated with subsequent progression to respiratory failure. The use of the so-called 20/30/40 rule allows patients-at-risk to be identified and to be preemptively admitted to the ICU for close monitoring and elective intubation under optimal conditions. In the past, clinical signs of respiratory fatigue and severe bulbar weakness with aspiration, or fall of the VC to less than 15 mL/kg or arterial PO2 values to less than 70 mmHg, had been defined as criteria for intubation (71). Although these criteria might be accurate in determining the need for intubation, they cannot be considered as practice guidelines for preemptive respiratory management of children with GBS. When serial bedside respiratory function testing shows constant decline of VC, PImax, PEmax, fulfilling criteria for the 20/30/40 rule, or more than 30% reduction in VC from baseline, the patient should be transferred to the ICU and elective intubation should be considered. It is also prudent to have young children with oxygen desaturation events with values falling below 90% and/or signs of dysautonomia, bulbar dysfunction, or aspiration transferred to the ICU for monitoring and possible intubation. One also needs to be aware of the fact that dysautonomia in GBS patients may also be triggered or accentuated by tracheal manipulation (i.e., intubation, suctioning). Because of the propensity of GBS patients for upper airway collapse, noninvasive methods of respiratory support such as continuous or bilevel positive airway pressure methods have probably a limited role in the respiratory management of severe GBS patients with borderline respiratory values. Nocturnal decompensation is also a common occurrence in patients with severe GBS due to a combination of reduction in VC related to the supine position and also impairment of central respiratory drive; thus the need for close respiratory monitoring during sleep. Immune therapies, plasma exchange, and high-dose intravenous immunoglobulin have been shown to be of equal efficacy and are known to expedite recovery from GBS (72).

Another major immediate diagnostic consideration in the differential diagnosis of GBS relates to excluding an acute spinal cord tumor or transverse myelitis. Children with either lesion may also be admitted to the medical ICU (73–76). Both of these disease entities may acutely produce a rapidly progressive paralysis, hyporeflexia, and back pain. More commonly spinal cord lesions, and rarely GBS, are associated with early sphincter dysfunction. However, in contrast to spinal lesions, these symptoms are usually transient with GBS (47). Four children had severe pain, asymmetric lower extremity weakness, and a clear-cut sensory level on careful neurologic examination (73). MRI was abnormal in four of the eight children evaluated (73). Each child had a malignant spinal cord tumor (73). Even imaging studies may cause confusion. One 6-year-old child developed progressive weakness and areflexia (74). Both the clinical and EMG findings were commensurate with a GBS diagnosis. If there is enlargement of the spinal cord, swelling with increased signal intensity of the cord, and no contrast enhancement, a diagnosis of GBS is not excluded because, rarely, transverse myelitis and GBS may occur concurrently (74,75). Transverse myelitis is the primary spinal cord lesion that may occasionally produce significant confusion in the differential diagnosis of GBS (76).
Case 9: Transverse Myelitis With Acute Gait Difficulty and Back Pain
A 12-year-old boy had sudden problems walking, tending to waddle with his knees flexed, the night before coming to BCH. Initially, his parents thought he was actually clowning. Later that night he developed knife-like back pain. In the morning he was found paraplegic and unable to urinate. Neurologic evaluation here demonstrated a flaccid paraplegia, absence of muscle stretch reflexes, and “ambiguous” responses to plantar stimulation. Arm and hand strength as well as their respective muscle stretch reflexes were normal. Sensory examination was somewhat limited in accuracy; there was a questionable sensory level at T10-12. CSF evaluation demonstrated 30 WBCs with 90% monocytes, and protein of 175 mg/dL. Spinal imaging demonstrated a tethered cord. No peroneal or tibial F waves were defined; routine peroneal, tibial, and median motor nerve conduction velocities (MNCVs) and sural SNAPs were normal. Needle EMG was unremarkable. MRI of the spinal cord showed T2 hyperintensities extending longitudinally in the lower thoracic region.
Comment
The acute onset of leg weakness, areflexia, and back pain with the absent F waves on EMG initially suggested a diagnosis of GBS with predominant involvement at the nerve root level. However, the lack of subsequent arm involvement, the persistent sphincter involvement, and the more precise definition of a saddle sensory level led to the appropriate clinical diagnosis of transverse myelitis. It is important to emphasize that although the absence of F waves is one of the most common early signs of GBS, similar findings are also found with transverse myelitis if the segmental lesion affects the precise spinal level for the legs or arms (47). EMG is generally normal in this setting, however, if segmental anterior-horn cells are affected, both the CMAPs and F waves may be affected. This leads to neurophysiologic mimicking of the typical early GBS findings. MRI usually confirms the diagnosis.
Mononeuritis Multiplex
During our 32-year experience at BCH, we have evaluated just one child with an acute fulminating mononeuritis multiplex (MNM) affecting a very ill teenager in renal failure. We have seen only two instances of MNM in more than 32 years at BCH. A colleague at Johns Hopkins recalls just one case (T. Crawford, personal oral communication, March 2002). This limited experience has been reported; two of the three cases were related to systemic vasculitis and one possibly represented a case of non-systemic vasculitis (77–79).
Case 10: Acute MNM
This 16-year-old girl with systemic lupus erythematosus had dialysis dependent, membranous, proliferative glomerulonephritis associated with hypertension, pericardial effusion, and seizures. She was not a renal transplant candidate because of medication noncompliance. A sudden onset of left-hand numbness occurred just 1 month before we initially evaluated her. She developed an acute left foot drop shortly thereafter followed by an acute numbness in her right hand and an acute right foot drop the very next day. She became incapacitated. Neurologic examination demonstrated a left median and ulnar and bilateral asymmetric peroneal neuropathies.
EMG demonstrated an MNM. Sural nerve biopsy confirmed the clinical diagnosis of a vasculitis. Treatment with intravenous solumedrol (30 mg/kg, maximum 1 g) per day for 5 days followed by oral prednisone (80 mg/d) was initiated. Within just a few days, she developed an acute gastrointestinal distress with upper gastrointestinal bleeding. Gastroscopy demonstrated an active bleeding site. A gastric mucosa biopsy also demonstrated a vasculitis.
Comment
MNM is very uncommon in children and adolescents. Interestingly, although MNM is commonly seen in diabetics, we are not aware of such complication in the pediatric age group. However, this single case emphasizes that if a child has an underlying connective tissue disorder and presents with an acute mononeuropathy, especially with no evidence of a mechanical reason, the possibility of a systemic vasculitis needs to be considered early on in the differential diagnosis. Today, children with renal compromise much less commonly develop a polyneuropathy (80).
Neuromuscular Junction Disorders in the Older Child
The differential diagnosis of acute childhood bulbar and generalized weakness requires consideration of a neuromuscular transmission defect. MG and the Miller-Fisher variant of GBS are the primary considerations. When MG occurs in children, it is typically quite similar to its presentation in adults. However, occasionally one may see some clinical variants that lead to an intensive care admission before the diagnosis of MG is even entertained.
Another very rare, often unidentified, and unsuspected disorder is tick paralysis. This biologic toxin is a rare and dramatic neuromuscular transmission defect that always needs consideration in the differential diagnosis of GBS or MG, particularly among children from the preschool ages forward (19,81–85). Each child with acute weakness requires careful inspection of the scalp to exclude this unusual diagnosis. The pathophysiology here is also most likely a defect in neuromuscular transmission although this hypothesis is difficult to prove with standard EMG techniques.
Case 11: Tick Paralysis Presenting as Acute Quadriparesis, Dysarthria, and Neck Muscle Weakness
This 3-year-old girl had a recent upper respiratory infection. The night prior to admission to BCH she stubbed her toe and later asked her dad to carry her to the bathroom. The next morning she was unable to move well and had to be carried downstairs. When her parents realized that she could not lift her head to watch television they took her to the local pediatrician. She found the child to have a weak voice, quadriparesis, areflexia, and normal sensation.
Careful inspection of the scalp demonstrated a tick. The pediatrician removed the tick and referred the child to BCH. The next morning this little girl had a very significant improvement. She was able to sit up with support and walk to the bathroom. Although intellectually of interest, we did not feel an EMG was needed with her rapid improvement.
Comment
One of the most helpful clinical clues, allowing the clinician to differentiate tick paralysis from GBS or MG, is the finding of early pupillary involvement, as seen in four of six Australian cases (19). Total ophthalmoplegia occurred in two patients and all but one of six children had extraocular muscle paresis. Those findings may sometimes suggest the Miller-Fisher GBS variant. Nonreactive pupils also occur with diphtheria and rarely GBS (58).
Typical EMG findings in tick paralysis include very diminished CMAP amplitudes with preserved MCVs, motor DLs, and SNAPs (19,84,85). These findings are similar to AMAN or even some early demyelinating forms of GBS. Sequential NCS studies immediately prior to removal of a tick, a few days hence, and in 6 months demonstrated that the reduced CMAP improved dramatically after the tick was removed (84,85). No neuromuscular transmission defect has been demonstrated with standard EMG RMNS (84,85). However, experimental data do point to tick paralysis being related to the presynaptic NMJ with decreased acetylcholine release (86). In some children the early appearance of fibrillation potentials, as soon as 24 to 48 hours, may lead to diagnostic confusion if one does not pay close attention to the total constellation of neurophysiologic findings (84,85).
Tick paralysis evolves more slowly than other biologic toxins, however, it may actually be more deadly (19). Prolonged respiratory paralysis occurred in two of the six Australian children (19). There the responsible tick Ixodes holocyclus is a different species than Dermacentor andersoni or Dermacentor variabilis common to North America (19). The latter is associated with a rapid improvement once the tick is removed in contrast to a continued deterioration for up to 48 hours in the Australian variety. In that setting the physicians must be careful not to discharge the child too soon. Four of their six children deteriorated after initial hospital discharge (19).
Case 12: Myasthenia Gravis With Acute Dysphagia, Drooling, and Ptosis
This infant girl was very healthy through age 15 months when she developed drooling, irritability, and inability to finish her bottles. Her pediatrician, noting a red throat, performed a throat swab that proved to be rapid streptococcal negative. During this procedure, she developed acute respiratory distress. After emergent admission to a local hospital she had an otolaryngology evaluation during which she was intubated. She was diagnosed with middle lobe pneumonia and treated with ceftriaxone. Her fever resolved but she had continued difficulty swallowing liquids. While taking oral medication she suddenly became limp and lethargic, sustaining a respiratory arrest. She responded to positive pressure ventilation and corticosteroids. An emergent transfer was made to BCH. Although successfully extubated she had continued difficulties swallowing. When this child was extubated 4 days later, she was then noted to have bilateral ptosis, bifacial weakness, frequent cough after eating, and was unable to walk.
Tensilon (edrophonium chloride) testing with a dose of 1.6 mgm produced a dramatic improvement in her ptosis and facial appearance. A subsequent EMG, with 3 Hz RMNS, demonstrated 38% to 45% decrements in the CMAPs. There was no repetitive component to her CMAPs. Her initial, commercially performed acetylcholine receptor antibody was negative; however, it was then definitely positive at the Mayo laboratories. She was treated with Mestinon (pyridostigmine bromide), in gradually increased dosages to 10-mg tid, with gradual but variable improvement of her neuromuscular findings. Interestingly, the ptosis and bifacial weakness were the last to resolve. A thymectomy was considered but with her excellent course it was decided to await a recurrence of her symptoms. The medication was withdrawn during the second 6 months of her illness. She has been asymptomatic now for a number of years.
Comment
Autoimmune MG (AMG) is a rare but important consideration in the differential diagnosis of either acute respiratory distress or failure to wean from a respirator. This applies to children of any age, even in toddlers, as noted in this instance. The youngest child with nonneonatal AMG that we are aware of was a 9-month-old (87). Although most AMG children present with ptosis and/or diplopia (88), we occasionally see other clinical settings, some of which require urgent intensive care management (89). This is particularly so when there is any hint of airway compromise. For instance, we evaluated one teenager being followed by an otolaryngologist for progressive but variable speech difficulties when suddenly she developed severe dysphagia and increasing respiratory distress. The reader is referred to Andrews’ (90) treatment outline.
■ MUSCLE
Channelopathies
Some of the various channelopathies, particularly hyperkalemic and hypokalemic periodic paralysis, always warrant consideration in the differential diagnosis of acute GBS. As these are autosomal dominant sodium or calcium channelopathies, there is usually a well-documented family history. Clinically the episodes of paralysis are relatively short-lived on most occasions. Routine NCS findings are normal; however, prolonged periods of exercise with EMG monitoring can evoke a diminution in CMAP amplitudes (91). Needle EMG often demonstrates significant myotonic-like discharges with the hyperkalemic variant.
We do call attention to a case of an 18-month-old, with a previously unrecognized sodium channel myotonia, who presented with respiratory stridor and increasing difficulty breathing, secondary to an acute bronchiolitis. She had trouble opening her eyes that was worsened with crying, as was her stridor. This child had somewhat elevated serum potassium presumably secondary to her increased respiratory effort. These findings were all compatible with a form of paramyotonia. This clinical combination exacerbated her underlying sodium channelopathy with vocal cord myotonia and led to ever increasing respiratory stridor.
Dermatomyositis
Very occasionally one may witness the acute onset of dermatomyositis in the pediatric age group. More commonly this illness has a slowly ingravescent temporal profile and does not require evaluation in the ICU.
■ CRITICAL ILLNESS NEUROMUSCULAR DISORDERS
The acute weakness or paralysis which may occasionally occur in children with critical illness is sometimes related to conditions involving the peripheral nerve, endplate, or muscle. Children with overwhelming sepsis or status asthmaticus may develop a critical illness neuromuscular syndrome that may mimic a hospital-acquired GBS presenting with a failure to wean from the respirator (92–94). Some of these syndromes had initially been felt to be secondary to a defect in neuromuscular transmission. Bolton was one of the first to emphasize the need for us to consider these various syndromes when we are called to the ICU (93). Subsequent to his initial report, defining the role of the peripheral nerve in some of these critical care failures to wean patients, further careful studies have led to the demonstration that most of these children may have a primary myopathy (95). These various critical illness neuromuscular disorders may affect each level of the motor unit (Table 21.4).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


