81 Neuromuscular Disorders
Neuromuscular disorders include a highly variable group of diseases that affect the peripheral nervous and muscular systems on any level of the neuraxis. Pathology ranges from disorders affecting the spinal motor neuron to the muscles by way of the peripheral nerves (Figure 81-1). In this chapter, several of the more common pediatric neuromuscular disorders are reviewed and classified based on their level of involvement in the neuraxis.
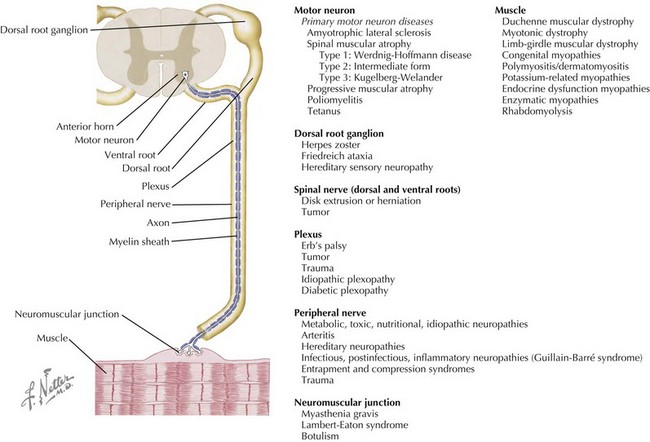
Anterior Horn Cell
Spinal Muscular Atrophy
Clinical Presentation
SMA is subclassified into four types based on age of onset and the maximal level of motor skills achieved. SMA type 1 (or Werdnig-Hoffmann disease) is the most common and severe of these disorders, accounting for approximately 60% of cases. SMA type 1 presents in the early infancy period (0–6 months) with generalized hypotonia, proximal and symmetric flaccid muscle weakness (initially lower more than upper limbs), and absent deep tendon reflexes. This frequently comes to the attention of the pediatrician with gross motor milestone delay and may present subacutely or more indolently. Most infants also have tongue fasciculations, and some have postural tremor of the fingers or joint contractures. Diaphragmatic sparing with intercostal muscle involvement results in paradoxical breathing and the classic bell-shaped torso (Figure 81-2). Importantly, these infants are alert and interactive with normal cognitive development and no sensory loss or impairment of eye movements. Systemic complications of SMA include pneumonia, scoliosis, poor weight gain, sleep difficulties, and joint contractures. These infants never achieve independent sitting. Most individuals’ expected life span is less than 2 years without invasive ventilatory and nutritional support.
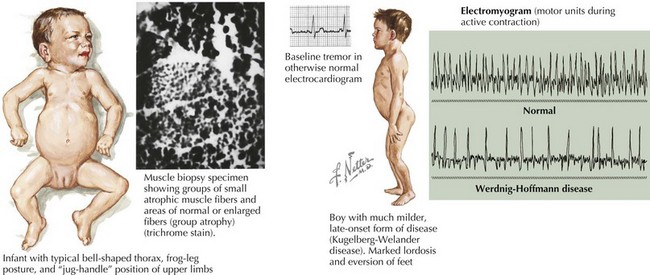
The differential diagnosis includes other disorders causing acute weakness, including poliomyelitis and infantile botulism. In addition, the general differential diagnosis for hypotonia and more chronic weakness in an infant should be considered (Table 81-1).
Table 81-1 Differential Diagnosis of the Floppy Baby, Infant, and Child
| Localization | Diagnoses (Examples) |
|---|---|
| Brain/Systemic | Chromosomal (Turner’s syndrome, trisomy 21, Prader-Willi syndrome) |
| Benign congenital hypotonia | |
| Infection (sepsis, meningitis, encephalitis, TORCH infections, tick paralysis) | |
| Metabolic (electrolyte abnormalities, hypothyroidism, hepatic encephalopathy, mitochondrial and peroxisomal disorders, amino and organic acidemias) | |
| Toxins (alcohol, narcotics, heavy metal poisoning, organophosphates, anticholinergics) | |
| Neonatal encephalopathy | |
| Trauma | |
| Spinal cord | Hypoxic-ischemic myelopathy |
| Compression | |
| Syringomyelia | |
| Anterior horn cell | Spinal muscular atrophy |
| Infection (polio, Coxsackie) | |
| Cytochrome C oxidase deficiency | |
| Peripheral nerve | Demyelinating (Guillain-Barré syndrome, hereditary motor-sensory neuropathy type I, congenital hypomyelinating neuropathy) |
| Axonal (familial dysautonomia, hereditary motor-sensory neuropathy type II, infantile neuronal degeneration) | |
| Neuromuscular junction | Infection (botulism) |
| Myasthenia gravis | |
| Muscle | Myopathies and congenital muscular dystrophies |
| Metabolic (acid maltase deficiency, hypo- or hyperthyroid myopathy, carnitine deficiency) | |
| Muscular dystrophies | |
| Inflammatory (dermatomyositis, polymyositis) | |
| Mitochondrial myopathies |
TORCH, toxoplasmosis or Toxoplasma gondii, other infections, rubella, cytomegalovirus, and herpes simplex virus.
Peripheral Nerve
Guillain-Barré Syndrome
Etiology and Pathogenesis
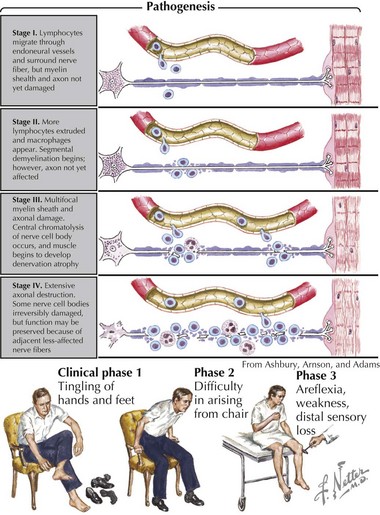
Pathologically, the two primary changes seen in GBS are acute inflammatory demyelinating polyradiculoneuropathy and acute axonal degeneration, both of which are thought to be caused by cross-reacting antibodies to the various gangliosides expressed in peripheral and cranial nerves. Campylobacter infections can result in molecular mimicry with the GM1 ganglioside, such that patients with C. jejuni infections are at 100-fold higher risk of developing GBS than the general population (Figure 81-3).
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


