15 Neurology
Neurologic Examination
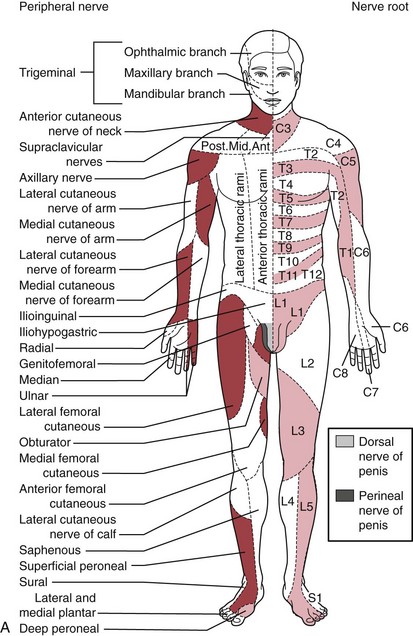
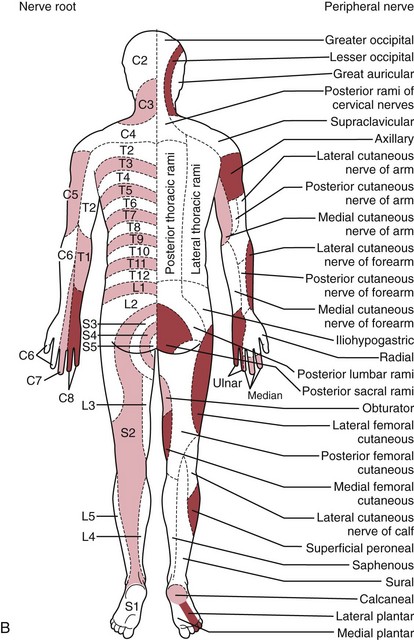
The traditional systematic neurologic evaluation proceeds from assessment of mental status and language functions through evaluation of cranial nerves, gross motor function, muscle strength, gait and station, balance and coordination, sensory systems, and deep tendon reflexes. It is applicable to older children and adolescents without significant modification from the evaluation geared to the adult. Tools essential to the neurologist include the reflex hammer, bright penlight, ophthalmoscope, and stethoscope. For evaluation of the primary sensory modalities of light touch, pain, temperature, and vibration, wisps of cotton, sterile disposable pins, glass test tubes (to hold hot and cold water), and a tuning fork (256 Hz for children and young adults, 126 Hz for older persons) are used. A collection of small, common objects (e.g., coins, buttons, keys) to be identified by feel alone are useful for assessment of cortical sensations of stereognosis. Derangements of primary sensory function may be present with lesions at the level of the nerve roots, plexuses, or peripheral nerves. If a particular area of decreased sensation is identified in part of a limb, careful delineation of its boundaries often suggests root (dermatomal), plexus, or peripheral nerve involvement (Fig. 15-1, A and B).
Much of the remainder of the neurologic examination also lends itself to play, and in the second stage of observation a more detailed assessment of mental status, language, handedness, and fine and gross motor skills is performed by engaging the child in play. A selection of rattles, keys, spinning and mechanical toys, dolls, cars, small blocks, noise makers, tennis balls, hand puppets, crayons, and picture books supplement the traditional instruments. If further observation of gait is necessary, the examiner can have the child walk to or with the parent. Children older than 4 years of age love to show what they can do when asked to walk on their heels or toes, hop, or do tandem gait along a line. Pat-a-cake games are popular for testing rapidly alternating movements with young children. Then, with the child comfortable and rapport established, the hands-on examination is initiated with the child still dressed and in the parent’s lap. Trying to catch the otoscope light as it is shown over various parts of the body can precede following the light with the eyes and looking at it. For infants and toddlers, following a face or spinning toy is still better for testing extraocular movements (Fig. 15-2). Having a parent jingle keys at the child’s eye level and asking the child to look at the sound while looking in the child’s eyes assists the ophthalmoscopic examination in older preschool and young school-age children (Fig. 15-3). Asking young children to make faces, stick out their tongues, and blow up balloons is another helpful technique in assessing cranial nerves.

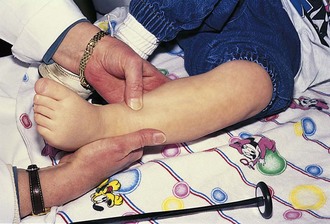
Tone is assessed by observing resistance to passive motion. Then active motion and motion against resistance are checked. Older preschoolers and school-age children love showing their muscles, and push–pull games can be used to test muscle strength, especially when the child’s efforts are admired. Deep tendon reflexes can often be tested at this time with only the shoes off. These are normally brisk, or 3+, in the young infant, becoming 2+ by 6 months of age. If directly tapping on the tendon seems upsetting to the child, it may help to place a finger over the tendon to be percussed and tap that. In infants and toddlers, it is often easier to elicit the ankle jerk by placing a finger over the ball of the child’s foot and gently dorsiflexing it before percussing the Achilles tendon (Fig. 15-4). Preschoolers and young children love having the examiner express surprise and pleasure when reflexes are elicited.
Finally the parent is asked to help undress the child, and the remainder of the examination proceeds with the parent providing reassurance and assistance as needed. During this stage, head circumference is measured in the infant and toddler, and the head, midline of the neck and back, and skin are carefully examined for abnormalities. Muscles are inspected for symmetry, and extremity circumference is measured a set distance from a bony landmark if asymmetry is suspected, and abnormal muscle movements are noted. The appropriate disappearance or persistence of primitive reflexes is determined in infants (see Chapter 3). The Babinski reflex is difficult to elicit and interpret during the first year of life because stroking the sole of the foot may simply stimulate withdrawal or plantar flexion. Using the Oppenheim technique—running the thumb down the medial surface of the tibia—gives a more interpretable response (Fig. 15-5). Evaluation of sensation is difficult in the younger child and is generally limited to appreciation of light touch and pinprick. These may be assessed with minimal discomfort by using a partially unbent paper clip.
Neurologic examination of the newborn is highly specialized. The essential components of the neonatal examination include assessment of gestational age, growth patterns, dysmorphic features, motor tone, postures, spontaneous activity, cry, respiratory patterns, brainstem reflexes, response to bright light, response to noxious stimuli, developmental reflexes, and deep tendon reflexes (see Chapter 2). Normal findings vary with gestational age. The immaturity of the newborn’s CNS, with functioning largely at a subcortical reflex level, may conceal all but the most severe neurologic deficits—hence the often deceptively normal examination of the newborn with hydranencephaly.
Neurocutaneous Syndromes
Neurofibromatosis 1
Neurofibromatosis 1, or NF-1 (previously known as von Recklinghausen disease), is the most common of the neurocutaneous syndromes and affects about 1 in 3000 individuals. Although usually inherited as an autosomal dominant disorder, up to 50% of cases may be sporadic. Molecular genetic testing for mutations of the NF1 gene is clinically available. The NF1 gene has been localized to chromosome 17q. Neurofibromin, its gene product, acts as a tumor suppressor, and its function is altered in affected patients. Characteristic clinical manifestations include multiple hyperpigmented skin macules (café-au-lait spots), axillary or inguinal freckling, multiple skin neurofibromas, and iris hamartomas (Lisch nodules). Associated abnormalities may include optic gliomas; other CNS tumors of glial or meningeal origin; neurofibromas of spinal or peripheral nerves; pheochromocytoma, macrocephaly, and cognitive impairment; and bony abnormalities. Diagnostic criteria for NF-1 are summarized in Table 15-1.
Table 15-1 Diagnostic Criteria for Neurofibromatosis 1
NF-1, neurofibromatosis 1.
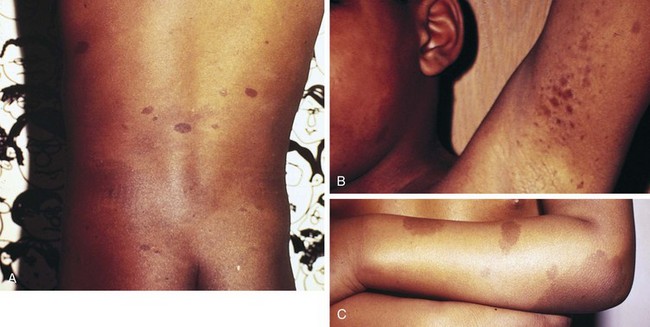
Multiple café-au-lait spots, the most frequently encountered cutaneous abnormality, are brown hyperpigmented macules with smooth margins, usually most numerous over the trunk (Fig. 15-6, A; and see Chapter 8, Fig. 8-131, A-C). Other abnormalities of cutaneous pigmentation may include axillary or inguinal freckling or extensive areas of hyperpigmentation (Fig. 15-6, B and C). Hyperpigmented skin lesions almost always precede neurologic symptoms and often increase in size and number with advancing age. They are not necessarily present at birth and may be inconspicuous in early childhood, becoming more prominent at puberty. Ninety-seven percent of patients with NF-1 have at least five café-au-lait spots by 20 years of age.
Although multiple café-au-lait spots are a clinical hallmark of NF-1, they may also occur as an autosomal dominant trait unassociated with the other features of neurofibromatosis. Genetic investigations in such families have excluded linkage to the NF1 locus on chromosome 17, indicating a distinctly different genetic association. Multiple café-au-lait spots can be found, as well, in a variety of other conditions (Table 15-2). They are a prominent feature of McCune-Albright syndrome, the additional manifestations of which include skeletal dysplasia and endocrine abnormalities. Those seen in McCune-Albright syndrome are often large and have irregular (“coast of Maine”) margins (see Chapter 8, Fig. 8-131, D), in contrast to the smooth (“coast of California”) borders characteristic of the hyperpigmented lesions of NF-1. Café-au-lait spots also may be encountered in tuberous sclerosis and neurofibromatosis 2 (NF-2) but are seldom prominent. They are present in about 10% of the general population (typically four or less in number) and are not by themselves a sign of disease.
Table 15-2 Conditions Associated with Café-au-lait Lesions
| Disorder | Clinical Features |
|---|---|
| Neurofibromatosis | Most common in NF-1, rare in NF-2 (see sections on NF-1 and NF-2 and Table 15-3) |
| Tuberous sclerosis | See Table 15-4 |
| McCune-Albright syndrome | Polyostotic fibrous dysplasia, precocious puberty, endocrine dysfunction |
| Watson syndrome | Features of Noonan syndrome (webbed neck, hypertelorism with antimongoloid slant, low-set ears, pulmonic stenosis, low intelligence), plus meets criteria for NF-1 |
| Epidermal nevus syndrome | Verrucous skin lesions, scoliosis, aortic coarctation, mental retardation |
| Bloom syndrome | Severe intrauterine and postnatal growth retardation, photosensitivity, telangiectatic erythema of cheeks/face |
| Ataxia-telangiectasia | Ataxia, conjunctival telangiectasia, recurrent sinopulmonary infections |
| Silver syndrome | Intrauterine growth retardation, hemihypertrophy, syndactyly, triangular facies, premature sexual development |
| Gaucher disease | Opisthotonos, splenomegaly, cranial nerve dysfunction, regression of motor milestones |
| Turner syndrome | Short stature, webbed neck, coarctation of aorta, delayed puberty |
| Fanconi anemia | Aplastic anemia, hyperpigmentation, short stature, mental retardation, congenital anomalies of bone, heart, eye, kidney |
| Multiple lentigines (LEOPARD) syndrome | Multiple small lentigines, hypertelorism, pulmonic stenosis, cryptorchidism, mild growth retardation, sensorineural deafness |
NF-1 and NF-2, neurofibromatosis types 1 and 2.
Additional cutaneous manifestations of NF-1 may include extensive plexiform neuromas at the terminal distribution of nerve fibers (Fig. 15-7; and see Chapter 8, Fig. 8-81) or small subcutaneous nodules—neurofibromas—scattered along the course of nerve trunks (Fig. 15-8).
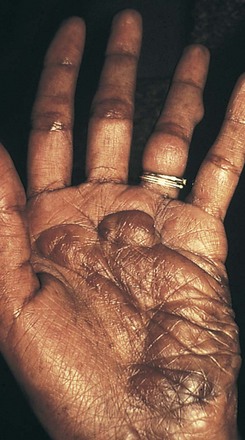
Figure 15-7 Neurofibromatosis 1 (NF-1). Extensive plexiform neurofibroma of the palm.
(Courtesy Michael Sherlock, MD, Lutherville, Md.)
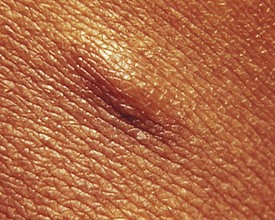
Figure 15-8 Neurofibromatosis 1 (NF-1). Subcutaneous neurofibroma along the course of a nerve trunk.
(Courtesy of Michael Sherlock, MD, Lutherville, Md.)
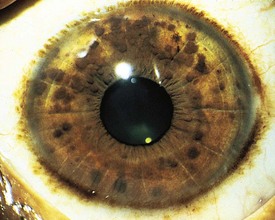
Pigmented hamartomas of the iris, termed Lisch nodules, are seen in more than 90% of patients with NF-1 who are older than age 6 years and can be found in nearly one third of younger individuals (Fig. 15-9). They do not occur in the normal population. Although these hamartomas are asymptomatic and do not correlate with the extent or severity of other manifestations, they are helpful in establishing the diagnosis.
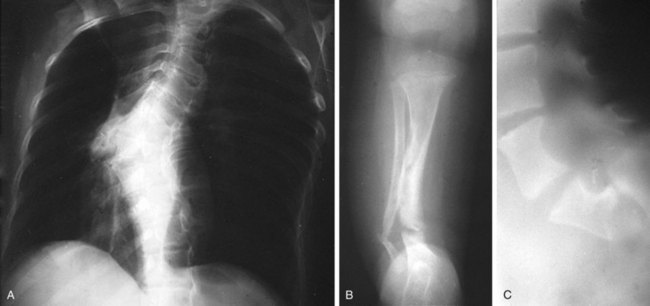
Short stature and macrocephaly are common in patients with NF-1, and skeletal abnormalities are found in 51% (Fig. 15-10). Osseous lesions, if present, usually appear in the first year of life. The characteristic findings include the following:
1 Severe angular scoliosis with dysplasia of the vertebral bodies (see Fig. 15-10, A)
2 Defects of the posterior superior wall of the orbit
3 Congenital bowing and thinning of the cortices of long bones and pseudarthrosis of the tibia, fibula (Fig. 15-10, B; and see Chapter 8, Fig. 8-131, B), femur, or clavicle (see Chapter 6, Fig. 6-77)
4 Disorders of bone growth associated with elephantoid hypertrophy of overlying soft tissue
5 Erosive bony defects produced by contiguous neurogenic tumors
6 Scalloping of the posterior margins of the vertebral bodies corresponding to saccular areas of dilation of the spinal meninges (Fig. 15-10, C)
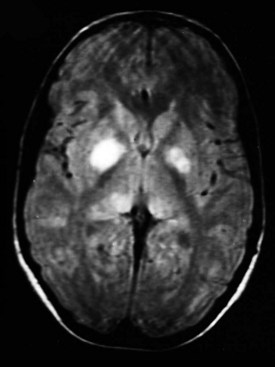
Magnetic resonance imaging (MRI) scans frequently show areas of increased signal intensity on T2-weighted images of the globus pallidus, brainstem, or cerebellar white matter (Fig. 15-11). These are commonly termed unidentified bright objects (UBOs) and are believed to represent increased fluid within the myelin associated with dysplastic glial proliferation. UBOs do not appear to correlate with neurologic dysfunction. However, their presence helps confirm the diagnosis of NF-1. Computed tomography (CT) seldom demonstrates corresponding abnormalities.
Neurofibromatosis 2
Symptoms usually first appear in the teens or early twenties, when pressure on the vestibulocochlear or facial nerve complex results in impaired auditory discrimination, hearing loss, tinnitus, unsteadiness, or facial weakness. Presenile lens opacities, found in half the patients examined, may precede the onset of symptoms referable to acoustic neuroma. Other Schwann cell tumors of cranial nerves, spinal roots, or the spinal cord, as well as multiple CNS tumors of meningeal or glial origin, may develop. Cutaneous manifestations such as café-au-lait spots, cutaneous neurofibromas, and axillary or inguinal freckling are less common in NF-2 than in NF-1, and Lisch nodules are not typical. Diagnostic criteria for NF-2 are summarized in Table 15-3.
Table 15-3 Diagnostic Criteria for Neurofibromatosis 2
CT, computed tomography; MRI, magnetic resonance imaging; NF-2, neurofibromatosis 2.
Tuberous Sclerosis
The more prominent features of this neurocutaneous disorder include seizures (96%), mental retardation (60%), intracranial calcification (49%), tumors of various organs (including the brain, heart, liver, and kidneys), and cutaneous lesions. Seizures are the most frequent presenting complaint. Clinical expression can be quite variable even among affected members of the same family. Diagnostic criteria are summarized in Table 15-4.
Table 15-4 Diagnostic Criteria for Tuberous Sclerosis
| Major Features |
| Minor Features |
| Definite Tuberous Sclerosis |
| Two major features or one major and two minor features |
| Probable Tuberous Sclerosis |
| One major plus one minor feature |
| Possible Tuberous Sclerosis |
| Either one major feature or two or more minor features |
Modified from Roach ES, Gometz MR, Northrup H: Tuberous Sclerosis Complex Consensus Conference: Revised clinical diagnostic criteria, J Child Neurol 13:624–628, 1998.
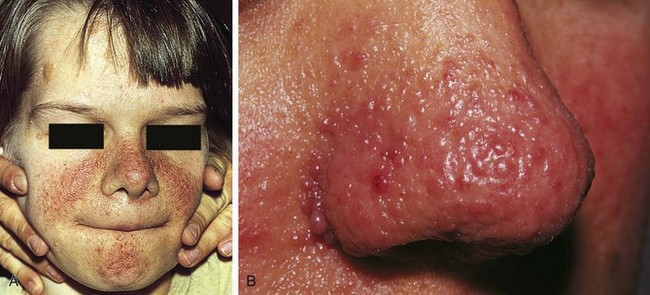
The characteristic skin lesion of tuberous sclerosis is the angiofibroma (adenoma sebaceum). These are seen as erythematous papules distributed over the nose and malar region of the face (Fig. 15-12). Approximately 40% of children with tuberous sclerosis demonstrate these lesions by 3 years of age.
Hypomelanotic macules with irregular borders, termed ash-leaf spots, are another common cutaneous manifestation (Fig. 15-13, A and B). These generally appear earlier than adenoma sebaceum and may be present at birth. They are detectable by 2 years of age in more than half of affected children. They resemble vitiligo but differ in that they are not completely devoid of melanin. In fair-skinned infants, these nevi may be demonstrable only under Wood lamp light.
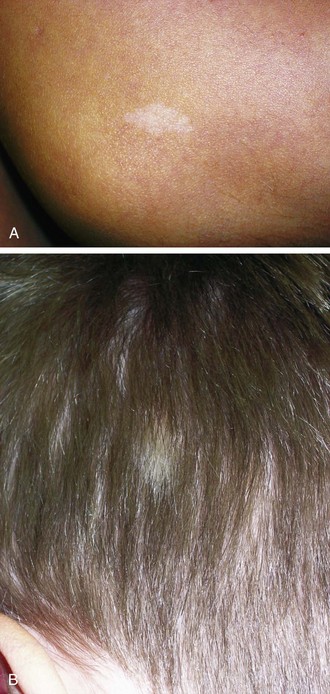
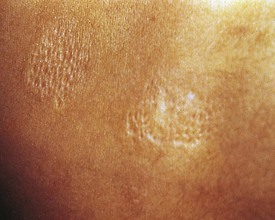
Another valuable cutaneous marker is the shagreen patch, a plaque of thickened skin with a cobblestone or orange-peel texture often seen on the dorsal aspect of the trunk (Fig. 15-14). Histologically, the shagreen patch is a connective tissue nevus.
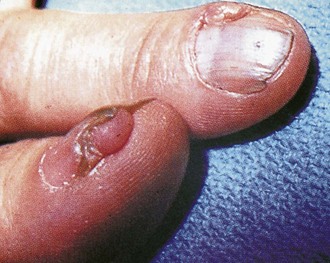
Additional dermatologic manifestations of tuberous sclerosis include periungual fibromas (Fig. 15-15) and macular areas of hyperpigmentation. Recognition of the cutaneous features can suggest an etiologic diagnosis in some patients with mental retardation or seizures. Oral examination may reveal pitting of dental enamel.
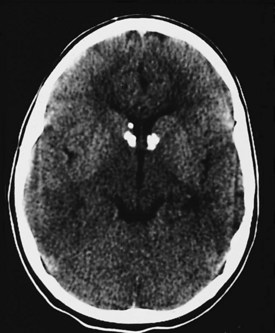
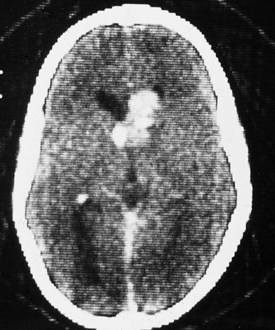
In patients with tuberous sclerosis, CT scans often demonstrate subependymal nodules seen as intracranial calcifications that appear as multiple scattered areas of increased density adjacent to the walls of the lateral and third ventricles (Fig. 15-16). CT is superior to MRI for demonstration of small calcifications. No relationship has been established between the extent of periventricular calcification and clinical severity as judged by developmental function or seizure frequency. CT may also demonstrate asymptomatic but typical intracranial calcifications in individuals who lack external manifestations of the disorder. This can help identify subclinical cases and improve the accuracy of genetic counseling in affected families.
The characteristic gross abnormality of the brain in TS is the presence of multiple gliotic nodules (hamartomas) of varying size, which constitute the tubers for which this disorder is named. These are located over the convolutions of the cerebral hemispheres and beneath the ependymal lining of the lateral and third ventricles. Heterotopic nodules of identical structure may be found in the cerebral white matter as well. Although cortical tubers are rarely apparent on CT scans, they are readily identified by MRI studies (Fig. 15-17). Severely affected patients have a greater number of cerebral cortical lesions detected by MRI scans, suggesting that MRI may be useful in predicting eventual clinical severity in young children with newly diagnosed tuberous sclerosis. Tumors may arise from cortical or subependymal tubers, complicating the course of the disease by producing increased intracranial pressure and other symptoms associated with intracranial mass lesions. Up to 80% of patients with tuberous sclerosis develop seizures of variable types that are often difficult to control. Infantile spasms are common and may be the presenting symptom leading to diagnosis. Patients with a history of infantile spasms and those who develop other types of seizures when very young tend to have more severe seizure disorders and poorer cognitive function.
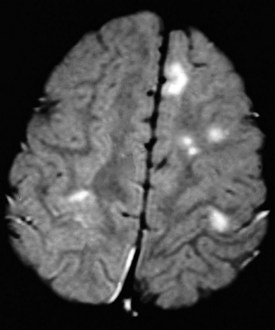
Subependymal giant cell astrocytomas (SEGAs) (Fig. 15-18) can affect 10% of patients with tuberous sclerosis and are clinically manifested by symptoms of obstructive hydrocephalus. The site of obstruction is often at the level of the foramen of Monro in the lateral ventricles. Such patients may present with signs of increased intracranial pressure (headache, vision changes, and/or papilledema), behavior change, or worsening seizure control.
Sturge-Weber Syndrome
The cardinal manifestations of Sturge-Weber syndrome are as follows:
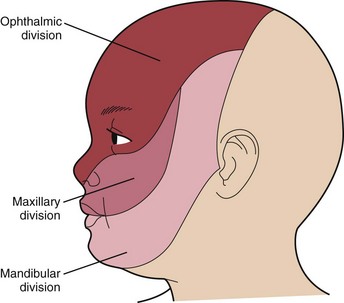
1 A vascular malformation or port-wine stain over the face that involves the cutaneous distribution of the ophthalmic division of the trigeminal nerve
2 Ipsilateral leptomeningeal angiomatosis with associated intracranial calcifications
3 A high incidence of mental retardation and ipsilateral ocular complications
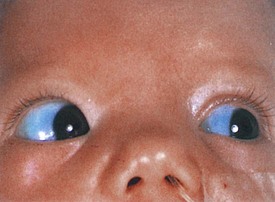
The port-wine stain (Fig. 15-19) is usually present at birth and consists of a pink-to-purple macular cutaneous vascular malformation. Only patients with lesions involving the cutaneous distribution of the ophthalmic division of the trigeminal nerve (i.e., forehead and upper eyelid) are at risk for associated neuro-ocular complications (Fig. 15-20). Repeated ophthalmologic and CT or MRI examinations are indicated only in this high-risk group, which has a 10% to 20% incidence of associated intracranial angiomas.
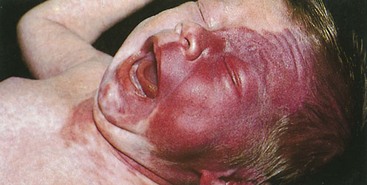
The coincidence of seizures and a facial vascular nevus should suggest the diagnosis of Sturge-Weber syndrome, which can be confirmed by CT scan (Fig. 15-21) or MRI. These scans may be normal at birth but subsequently show areas of gyriform contrast enhancement corresponding to the leptomeningeal angiomatosis. Serial examinations often demonstrate progressive ipsilateral cerebral atrophy. Additional findings may include serpiginous calcifications of brain parenchyma underlying vascular malformations of the pia. These intracranial calcifications are first seen on CT scan but become evident on plain skull films by the end of the second decade.
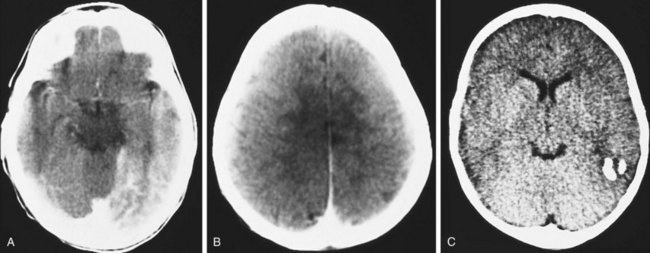
Associated ocular abnormalities are often encountered. Buphthalmos (corneal enlargement) or a coloboma may be present at birth, and glaucoma frequently develops in infancy or later childhood (Fig. 15-22). Dilated vessels in the sclera, conjunctiva, and retina are common, and angiomatous malformations of the choroid occasionally occur.
Klippel-Trénaunay Syndrome
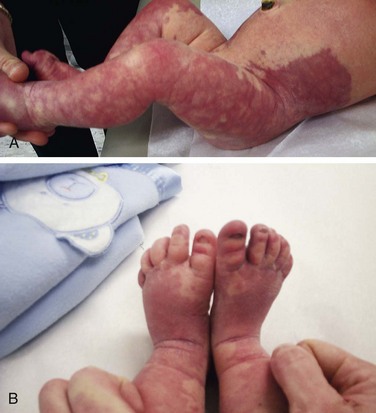
Patients with Klippel-Trénaunay syndrome (KTS) are born with a port-wine stain that is usually located over the lateral aspect of one leg. Less often an arm and a leg may be affected. In rare instances more extensive, even bilateral, involvement is seen. The surface lesion is associated with an underlying vascular malformation, which provides an unusually rich blood supply to soft tissue and bony structures that results in hypertrophy (usually hemihypertrophy) and lymphedema (Fig. 15-23, A and B). These features are often evident in the newborn, and progressive enlargement occurs during the first few years. Most cases are sporadic. However, a few cases have been reported to be autosomal dominant.
Ataxia-Telangiectasia
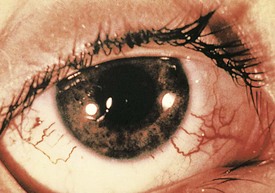
The characteristic cutaneous manifestations of this disorder appear by 6 years of age. Telangiectases first appear on the bulbar conjunctivae (Fig. 15-24) and develop later over the malar regions, ears, antecubital fossae, neck, and upper chest.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


