Danielle R. Rios, Angelica Vasquez, Christopher McPherson Key points There are a number of cardiovascular medications that are used in neonatology on a regular basis. An understanding of physiology can guide the clinician toward the selection of a specific medication based on mechanism of action that is necessary to manage the patient-specific disease state. Each medication will have a range of side effects that should be considered when choosing a medication therapy regimen, and even more so when utilizing this class of medications in critically ill neonates. Extrapolating data from older pediatric or adult patients must be done with caution considering the unique physiology of critically ill neonates. For example, the premature neonatal heart contains only 30% contractile tissue, in contrast to 60% in the adult heart.1 Additionally, expression of sarcoplasmic reticulum and t-tubules is low, while mitochondria are abundant, resulting in disorganized myocyte activity. These maturational differences are tolerated in the intrauterine environment dominated by low placental resistance. However, with exposure to ex utero systemic vascular resistance, the stress of illness, and inotropes or vasopressors, cardiovascular function may be impaired. The adrenergic system and myocardial innervation both mature throughout gestation.2 This maturation is driven to some extent by stimulation, and fetal adrenoreceptors have a low threshold for provocation. In preterm neonates this manifests as “denervation hypersensitivity,” in which myocardial adrenoreceptors demonstrate maximal response to even small concentrations of catecholamine.3 Importantly, maturation is not uniform, as active alpha-1-receptors outnumber beta-1-receptors in early gestation.4 Consequently, agents with non-specific activity like dopamine have a different dose-response profile in premature neonates, term neonates, and older patients. Critical illness further complicates dose-response. Renal maldevelopment or maladaptation has implications on drug elimination with lower albumin binding leading to increased drug available for metabolism in the setting of less efficient renal clearance of active drug and metabolites. Hypoxic-ischemic encephalopathy complicates the consequences of bradycardia (induced by therapeutic hypothermia or medications like dexmedetomidine in the setting of existing cardiac dysfunction induced by hypoxia-ischemia) and tachycardia (induced by exogenous catecholamines in the setting of metabolic insufficiency and/or hypocalcemia compromising right ventricular performance).5 Additionally, decreased renal clearance in this population may exacerbate the therapeutic or adverse effects of pharmacologic interventions.6 In confluence, this complex milieu highlights the vital nature of optimizing diagnostic techniques and understanding the specific impacts of available pharmacotherapies. This chapter will discuss the mechanism of action and therapeutic effects of cardiovascular medications used most commonly in critically ill neonates and infants based on predominant pathology grouping (Tables 5.1 and 5.2). We will not be discussing medication regimens for treatment of a hemodynamically significant patent ductus arteriosus (PDA), as these are discussed in detail in Chapter 17. Critical evaluation of available pharmacologic studies and randomized controlled trials was vital to generation of the content of this chapter. However, this evaluation highlighted the profound limitations of the available data. Therefore the information in this chapter should be interpreted in the context of emerging evidence. 2–20 mcg/kg/min Alpha-2 effects less prominent 0.01–0.5 mcg/kg/min PDE-3 inhibitor ↓ SVR, PVR, inotropy, and lusitropy 0.25–1 mcg/kg/min PDE-3 inhibitor ↓ SVR, PVR, inotropy 0.05–0.2 mcg/kg/min Arterial and venous vasodilator 0.5–2 mcg/kg/min Selective pulmonary vasodilator ↓ PVR 1–20 ppm PDE-5 inhibitor Continuous IV: 0.4 mg/kg over 3 hours, then 1.6 mg/kg/day ETA and ETB blocker ↓ PVR 1–2 mg/kg BID Pulmonary and systemic vasodilator ↓ PVR, SVR Continuous IV: 50–80 ng/kg/min Analogue of epoprostenol ↓ PVR, SVR IV or subcutaneous infusion 2–20 ng/kg/min Analogue of epoprostenol ↓ PVR, SVR Inhalation: 0.25–2.5 mcg/kg q2–6 Vasodilation of vascular and PDA smooth muscle 0.01–0.1 mcg/kg/min sGC stimulation ↓ PVR 0.5–2 mg up to TID 0.1–1.2 mU/kg/min (0.006–0.072 U/kg/h) 0.02–1 mcg/kg/min 2–20 mcg/kg/min ↑ SVR 0.1–0.5 mcg/kg/min Variable ↓ SVR 0.05–0.15 mg/kg QID ↓ SVR 0.05–0.3 mg/kg 1–2 times daily ↓ SVR PO: 0.04–0.3 mg/kg/day div 1–2 times daily When attempting to support heart function, a combination of inotropes and vasodilators may be utilized. Inotropes are medications that act primarily on the cardiac myocyte (Figure 5.1) to increase contractility, whereas vasodilators decrease wall tension leading to an improvement in stroke volume and cardiac output. In this section we will discuss two predominant inotropes, dobutamine and epinephrine; two inotropes with vasodilator effects, milrinone and levosimendan; and sodium nitroprusside, a primary vasodilator. The major goal of medication therapy in PH in the acute setting is to improve pulmonary blood flow, and, more often than not, fast-acting medications are utilized first-line (e.g., pulmonary vasodilator). In addition, there are likely to be secondary beneficial effects to RV function and cardiac output. In the chronic setting, while fast-acting agents may be utilized initially, more often than not, chronic medication therapy is required to affect pulmonary pressures. In both cases, evaluation, and support, when decreased, of both RV and LV heart function is still required. Systemic hypotension is a common diagnosis in the neonatal intensive care unit (NICU), though its frequency varies considerably between institutions. The choice of medication will vary based on the presence or absence and degree of systemic perfusion, cerebral blood flow, heart function, pulmonary or systemic vascular resistance, and intra- or extra-cardiac shunt flow and direction. Commonly used medications in this setting include predominant systemic vasoconstrictors (i.e., dopamine, norepinephrine, vasopressin, and phenylephrine) and glucocorticoids.
Chapter 5: Neonatal cardiovascular drugs
Introduction
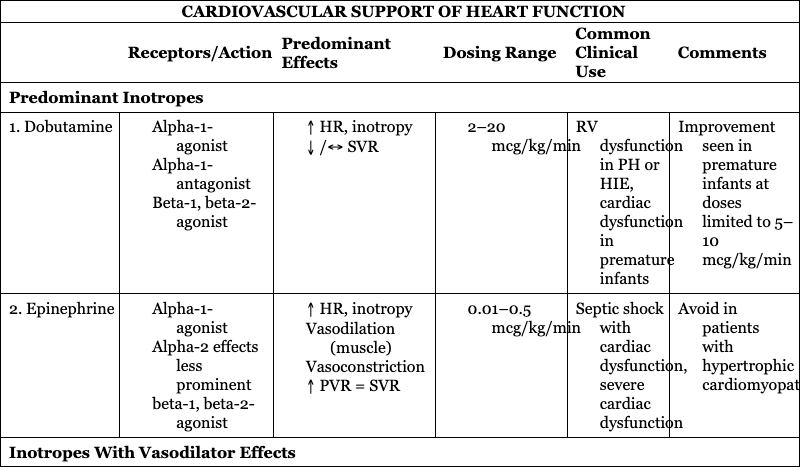
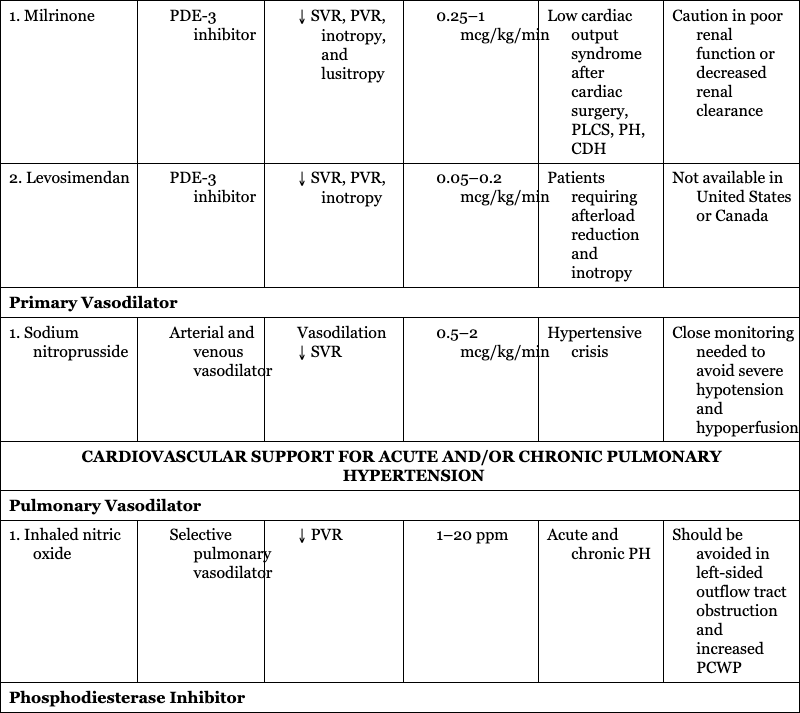


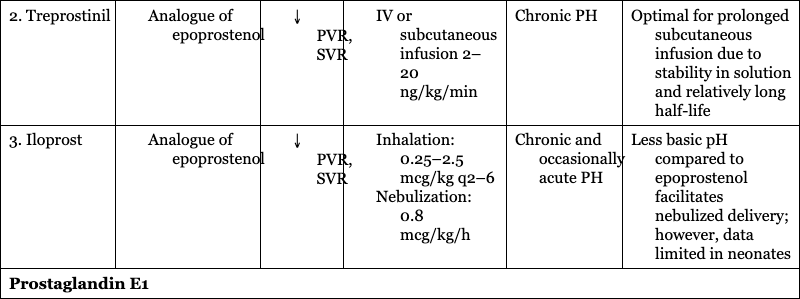
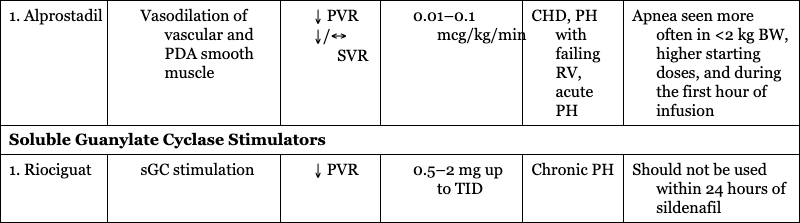
CARDIOVASCULAR SUPPORT OF HEART FUNCTION
Receptors/Action
Predominant Effects
Dosing Range
Common Clinical Use
Comments
Predominant Inotropes
1. Dobutamine
RV dysfunction in PH or HIE, cardiac dysfunction in premature infants
Improvement seen in premature infants at doses limited to 5–10 mcg/kg/min
2. Epinephrine
Septic shock with cardiac dysfunction, severe cardiac dysfunction
Avoid in patients with hypertrophic cardiomyopathy
Inotropes With Vasodilator Effects
1. Milrinone
Low cardiac output syndrome after cardiac surgery, PLCS, PH, CDH
Caution in poor renal function or decreased renal clearance
2. Levosimendan
Patients requiring afterload reduction and inotropy
Not available in United States or Canada
Primary Vasodilator
1. Sodium nitroprusside
Hypertensive crisis
Close monitoring needed to avoid severe hypotension and hypoperfusion
CARDIOVASCULAR SUPPORT FOR ACUTE AND/OR CHRONIC PULMONARY HYPERTENSION
Pulmonary Vasodilator
1. Inhaled nitric oxide
Acute and chronic PH
Should be avoided in left-sided outflow tract obstruction and increased PCWP
Phosphodiesterase Inhibitor
1. Sildenafil
Chronic and occasionally acute PH
Could contribute to worsened V:Q mismatch
Endothelin Receptor Antagonist
1. Bosentan
Chronic PH
Liver enzymes need to be followed closely
Prostacyclin Analogues
1. Epoprostenol
Chronic PH and elevated PVR
Abrupt discontinuation results in rebound PH
2. Treprostinil
Chronic PH
Optimal for prolonged subcutaneous infusion due to stability in solution and relatively long half-life
3. Iloprost
Chronic and occasionally acute PH
Less basic pH compared to epoprostenol facilitates nebulized delivery; however, data limited in neonates
Prostaglandin E1
1. Alprostadil
CHD, PH with failing RV, acute PH
Apnea seen more often in <2 kg BW, higher starting doses, and during the first hour of infusion
Soluble Guanylate Cyclase Stimulators
1. Riociguat
Chronic PH
Should not be used within 24 hours of sildenafil

SUPPORT FOR SYSTEMIC HYPOTENSION
Receptors/Action
Predominant Effects
Dosing Range
Common Clinical Use
Comments
Predominant Vasoconstrictors
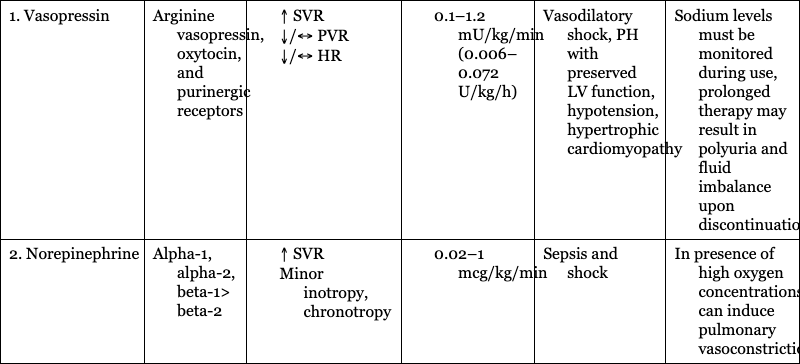
1. Vasopressin
Arginine vasopressin, oxytocin, and purinergic receptors
Vasodilatory shock, PH with preserved LV function, hypotension, hypertrophic cardiomyopathy
Sodium levels must be monitored during use, prolonged therapy may result in polyuria and fluid imbalance upon discontinuation
2. Norepinephrine
Alpha-1, alpha-2, beta-1> beta-2
Sepsis and shock
In presence of high oxygen concentrations, can induce pulmonary vasoconstriction
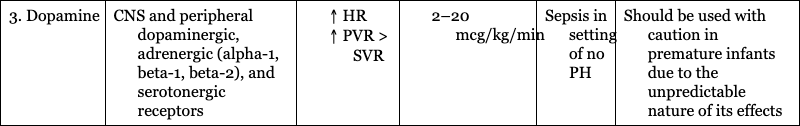
3. Dopamine
CNS and peripheral dopaminergic, adrenergic (alpha-1, beta-1, beta-2), and serotonergic receptors
Sepsis in setting of no PH
Should be used with caution in premature infants due to the unpredictable nature of its effects
4. Phenylephrine
Alpha-agonist
“Tet spells”
May cause severe bradycardia
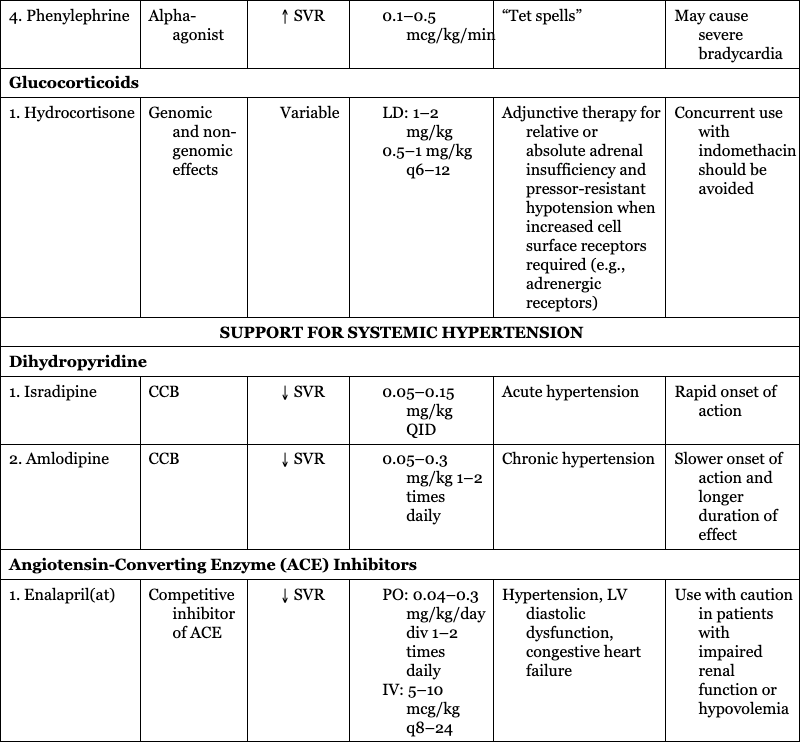
Glucocorticoids
1. Hydrocortisone
Genomic and non-genomic effects
Adjunctive therapy for relative or absolute adrenal insufficiency and pressor-resistant hypotension when increased cell surface receptors required (e.g., adrenergic receptors)
Concurrent use with indomethacin should be avoided
SUPPORT FOR SYSTEMIC HYPERTENSION
Dihydropyridine
1. Isradipine
CCB
Acute hypertension
Rapid onset of action
2. Amlodipine
CCB
Chronic hypertension
Slower onset of action and longer duration of effect
Angiotensin-Converting Enzyme (ACE) Inhibitors
1. Enalapril(at)
Competitive inhibitor of ACE
Hypertension, LV diastolic dysfunction, congestive heart failure
Use with caution in patients with impaired renal function or hypovolemia
Cardiovascular support of heart function
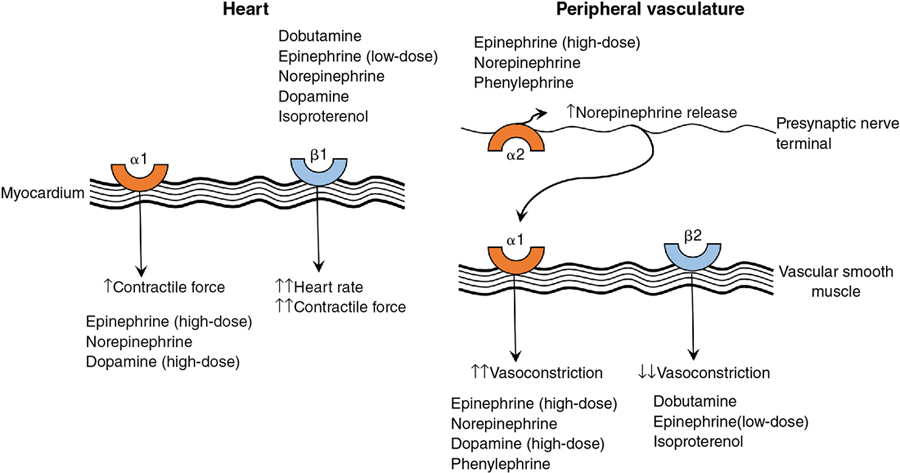
Predominant inotrope
Dobutamine
Epinephrine
Inotropes with vasodilator effects
Milrinone
Levosimendan
Primary vasodilator
Sodium nitroprusside
Cardiovascular support for acute and/or chronic pulmonary hypertension (Figure 5.2)
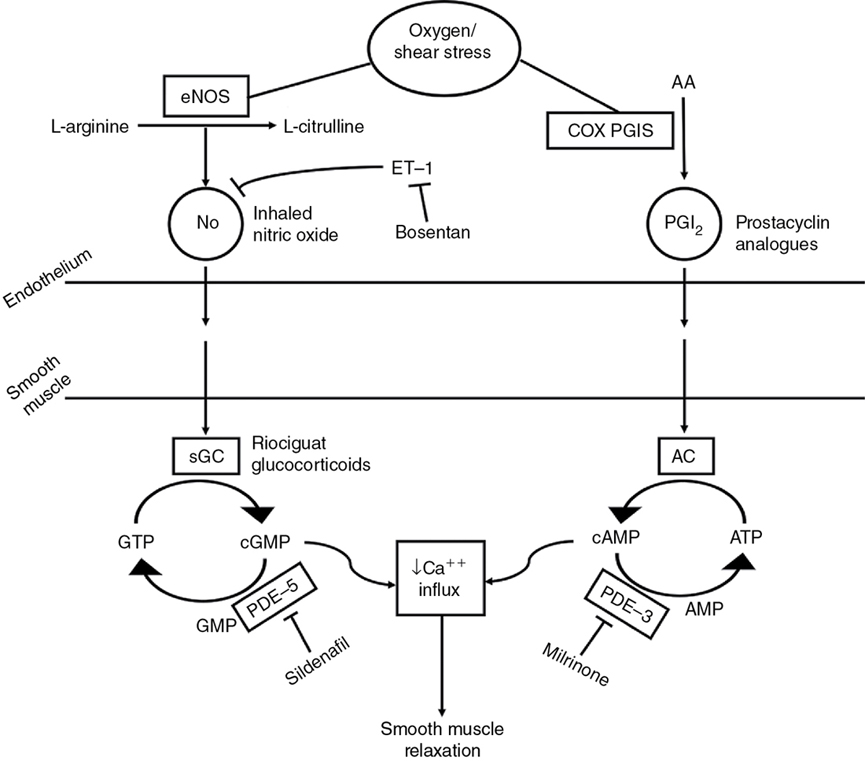
Pulmonary vasodilator
Inhaled nitric oxide
Phosphodiesterase inhibitors
Sildenafil
Endothelin receptor antagonists
Bosentan
Prostacyclin (analogues)
Epoprostenol
Treprostinil
Iloprost
Prostaglandin E1
Alprostadil
Soluble guanylate cyclase stimulators
Riociguat
Support for systemic hypotension
Predominant vasoconstrictors
Dopamine
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Obgyn Key
Fastest Obstetric, Gynecology and Pediatric Insight Engine
