In developing countries a simplified dosing schedule for gentamicin based on extended intervals saves time. Studies in Bangladesh and India established a suitable regimen of a fixed dose of 10 mg 48 hourly for infants <;2000 g, 10 mg 24 hourly for 2000–2500 g and 13.5 mg 24 hourly for infants >2500 g (Table 24.1),89,90 which has been evaluated and found to be safe.91
Table 24.1 Recommended antibiotic doses and intervals.
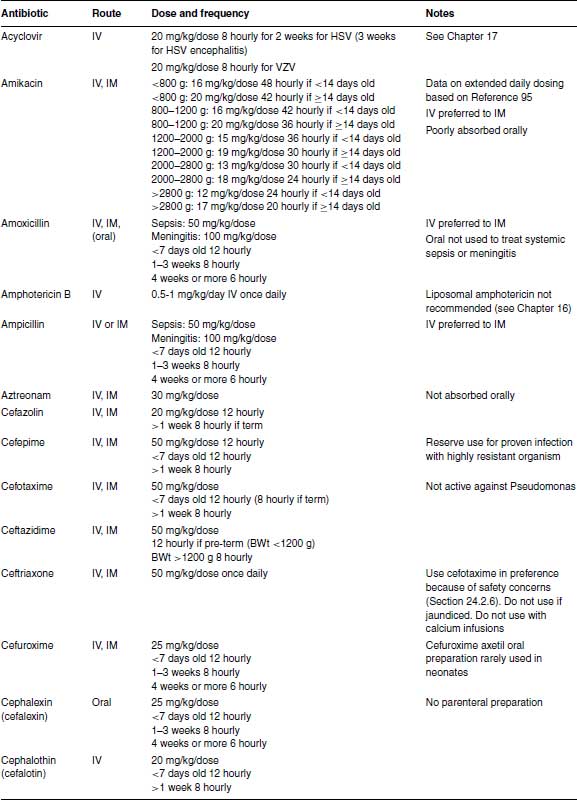
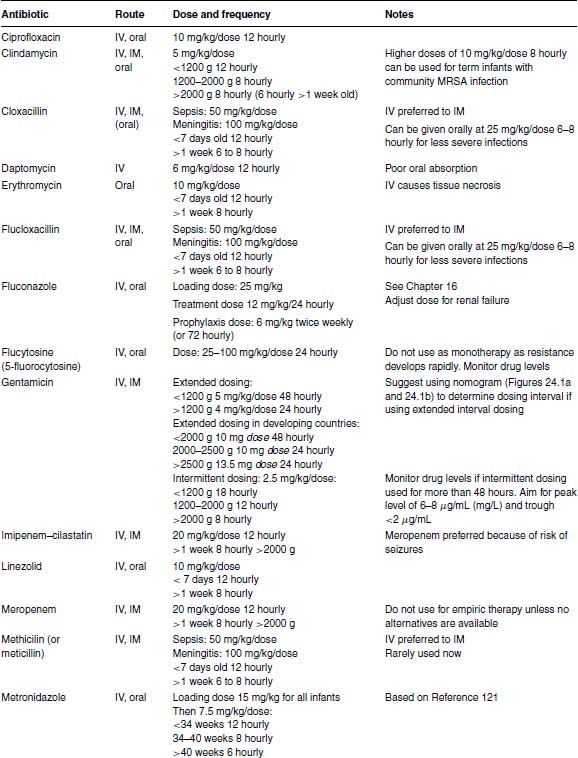
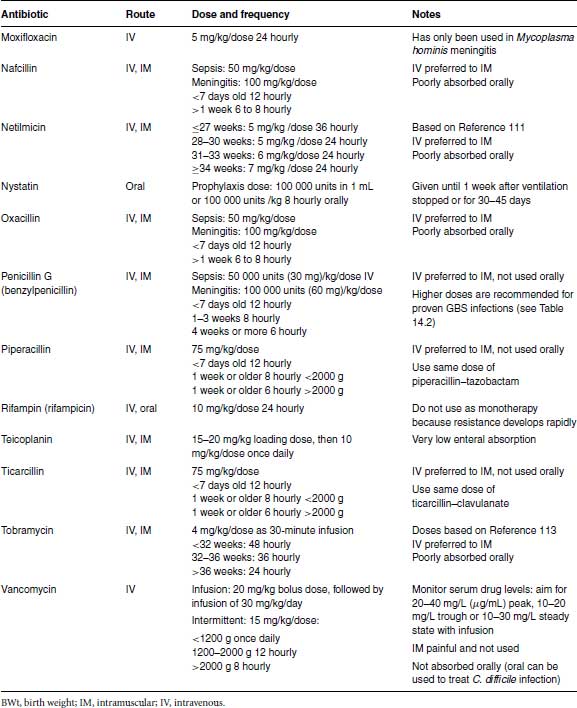
The initial doses for intermittent dosing of gentamicin are given in Table 24.1 for clinicians who prefer to stay with intermittent dosing until further data are available. If intermittent dosing is used, it is unnecessary to measure serum gentamicin levels (except possibly for medicolegal purposes) if gentamicin is stopped after 48 hours. However, if gentamicin is continued >48 hours, therapeutic drug monitoring is essential to minimize toxicity and ensure adequate therapeutic levels. The peak level taken 30–60 minutes after a dose should be 6–8 μg/mL (6–8 mg/L), while the trough level taken just before a dose should be <;2 μg/mL (>2 mg/L).
24.9.2 Amikacin
Amikacin is a semi-synthetic derivative of kanamycin A (which comes from Streptomyces kanamyceticus). Amikacin is relatively resistant to degradation by plasmid-mediated enzymes that mediate resistance to gentamicin and tobramycin. Some North American neonatal units wisely refrain from using amikacin, to ensure amikacin can be reserved for empiric treatment if widespread resistance to other aminoglycosides becomes a problem. In many developing countries widespread aminoglycoside resistance is indeed a major problem and amikacin is used commonly.92,93 However, resistance can emerge during treatment with P. aeruginosa and Serratia so amikacin should be used responsibly. In particular, it should be stopped if systemic cultures are negative and not continued because of concerns about resistance and about cumulative toxicity.
Amikacin can cause oto- and nephrotoxicity. Amikacin clearance depends on gestational age and post-natal age and is reduced in intrauterine growth retardation.94,95 Small studies did not show any toxicity with once-daily compared with intermittent dosing of amikacin,96–98 but too few infants were studied to guarantee its safety. Different dosing regimens have been proposed 99 but the largest pharmacokinetic modelling study which used data from 874 neonates concluded that previous dosing regimens might increase toxicity and developed a new model-based dosing regimen (Table 24.1).95
24.9.3 Netilmicin
There have been a number of pharmacokinetic and clinical studies on neonates using netilmicin.100–112 The conclusion of these is that renal clearance depends on gestational age, post-natal age and renal function, that the half-life is long and there is tissue accumulation as with gentamicin 100 and that extended daily dosing is safe and effective.107–112
24.9.4 Tobramycin
Tobramycin has slightly greater activity against P. aeruginosa than gentamicin in vitro but less activity against other organisms and is less nephrotoxic in animal models although this has not been shown in human neonates. Extended-interval dosing appears to be safe and achieves predicted levels.113 There is less experience with tobramycin in neonates than there is with gentamicin and netilmicin.
24.10 Quinolones
The quinolones are synthetic antibiotics based on a 4-quinolone nucleus and related to the parent drug nalidixic acid which was first used clinically in 1962. They act by interfering with DNA synthesis and are mostly well absorbed orally. The fluoroquinolones developed in the 1980s have improved activity against Enterobacteriaceae, Pseudomonas and gram-positive organisms compared with older quinolones. Newer agents marketed as ‘respiratory quinolones’ (such as moxifloxacin and gatifloxacin) have better anti-streptococcal activity than ciprofloxacin but lack reliable anti-pseudomonal coverage.
Several quinolones have been withdrawn because of toxicity, some are restricted to veterinary use and their use may contribute to quinolone resistance, while a number are useful human antibiotics.
Resistance occurs less with fluoroquinolones but is a problem particularly with prolonged use. The usual mechanism is chromosomal mutation in Enterobacteriaceae and efflux/porin loss in pseudomonads.
24.10.1 Ciprofloxacin
Ciprofloxacin is active against a range of gram-negative and gram-positive pathogens (although its anti-streptococcal activity is unreliable) and it is the only effective oral agent against P. aeruginosa. However, there are considerable concerns about toxicity: ciprofloxacin damages cartilage in beagle puppies and is associated with Achilles tendon rupture and retinal detachment in adult humans. It has been used in neonates but a systematic review reported only observational cohort studies, case series and case reports detailing its use as salvage therapy for sepsis caused by multi-resistant organisms.114 Ciprofloxacin was always started intravenously using various dosing schedules. Clinical response was estimated at 64% and 91% in two cohort studies, with a median of 83% in case series. Of the 14 case reports, 12 yielded positive clinical outcomes. No joint toxicity or other serious adverse events were reported on limited clinical evaluation and follow-up.114
Ciprofloxacin pharmacokinetic studies suggest that levels are similar for infants <;1500 and >1500 g and for infants <;7 and >7 days old.115 Ciprofloxacin has moderate CSF penetration and has been used as a last resort to treat meningitis 116–118 including P. aeruginosa meningitis and ventriculitis.118 Ciprofloxacin does not affect Pseudomonas colonization and should not be used to treat colonization with P. aeruginosa but should only be used to treat proven infection with multi-resistant organisms for which no alternative is available. Ciprofloxacin should not be used for prolonged periods because of the dangers of resistance and toxicity.
24.10.2 Moxifloxacin
Moxifloxacin is a recently developed quinolone with enhanced activity against gram-positive cocci, some gram-negative bacilli, including Acinetobacter, Aeromonas and S. maltophilia, and against anaerobes. It is less active than ciprofloxacin against Pseudomonas and other non-fermenting gram-negative bacilli. It has good CSF penetration and has been used together with a tetracycline (minocycline 119 or doxycycline 120) to treat Mycoplasma hominis meningitis. In one case report moxifloxacin 13 mg/kg/day was given orally,119 while a second case report used 5 mg/kg IV once daily successfully and reported the pharmacokinetic profile which showed lower serum levels than for an equivalent dose in adults.120
24.11 Metronidazole
Metronidazole is a nitroimidazole, which are agents isolated from streptomycetes with an unusual spectrum of activity against anaerobes, some microaerophilic species and protozoa including Giardia and Entamoeba histolytica. Metronidazole pharmacokinetics in neonates is best described using a one-compartment model with post-menstrual age as the main co-variate.121,122 A loading dose of 15 mg/kg is recommended 122 and has been predicted to give adequate serum and tissue levels for 24 hours for a pre-term infant and 48 hours for a full-term infant.121 Metronidazole has been used to provide anaerobic cover, for example, it is often used in Europe with other antibiotics in treating necrotizing enterocolitis (see Section 11.1.6), and has been used to treat rare cases of B. fragilis meningitis.123,124
24.12 Rifampin (rifampicin)
Rifampin (called rifampicin in Western Europe and Australia) is a member of the rifamycin family which are semi-synthetic antibiotics modified from rifamycin B which is produced by an actinomycete. Rifampin is mainly active against gram-positive organisms: S. aureus including MRSA and pneumococci including penicillin-resistant pneumococci, although enterococci are relatively resistant. Rifampin is active against gram-positive bacilli including Listeria, Bacillus, Clostridium and Corynebacteria. It has little activity against gram-negative enteric bacilli but is active against Neisseria species including meningococcus, H. influenzae, and Bacteroides. Rifampin is active against Mycobacterium tuberculosis (TB) and some atypical mycobacteria, but not against the Mycobacterium avium complex or M. fortuitum.
Rifampin is well-absorbed and can be given orally or intravenously, although intravenous administration is more likely to cause hepatotoxicity. It is well tolerated, but is a potent inducer of hepatic P450 microsomal enzymes and can enhance metabolism and clearance of other drugs that are metabolized in the same way.
Resistant mutations occur naturally in large bacterial populations and are rapidly selected by rifampin monotherapy which is, therefore, not advised. The only situations where rifampin monotherapy is useful are as short-term prophylaxis for carriers of Neisseria meningitidis or H. influenzae or as chemoprophylaxis for latent TB: in both situations there are such low numbers of organisms that resistance is not a problem. Rifampin is used in the treatment of congenital 125 and neonatal TB,126 is sometimes used as chemoprophylaxis for latent TB 126 and is sometimes used in conjunction with one or more agents to treat MRSA and other staphylococcal infections. One advantage is the ability to continue long-term oral therapy, for example, in treating osteo-articular infections if this route is preferred.
24.13 Antifungals
24.13.1 Amphotericin B
Amphotericin B is a polyene antibiotic derived from Streptomyces nodosus. Polyenes act by binding to sterols in the fungal cell wall. Amphotericin B is active against most of the organisms likely to cause neonatal infection including most strains of Candida, Aspergillus, zygomycetes and dermatophytes. Some strains of Candida parapsilosis are resistant and, after prolonged therapy, resistance may develop in C. lusitaniae and rarely in strains of C. guillermondii, C. krusei and C. tropicalis.
The main toxicity is renal, but amphotericin B can also cause fever and infusion reactions, although these are rare in neonates. As described in Chapter 16 (see Section 16.5.1) amphotericin B is relatively well tolerated in the neonatal period and renal toxicity is reversible and rarely necessitates changing to a different antifungal agent.127–129
Lipid-associated preparations of amphotericin B (liposomal amphotericin, amphotericin B colloidal suspension and amphotericin lipid complex) were developed to reduce toxicity and to improve delivery of amphotericin B to aqueous compartments such as CSF. A Cochrane systematic review of four trials comparing lipid formulations of amphotericin with conventional amphotericin B found reduced systemic and nephrotoxicity with lipid formulations but no improvement in outcome (mortality of complete resolution of fungal infection).130 Despite initial enthusiasm, there is now considerable concern that lipid formulations may actually be inferior to conventional amphotericin B in neonates. The mortality in the United States was 29% for infants with invasive Candida infections receiving liposomal amphotericin compared with 18% for conventional amphotericin (OR 2.0, 95% CI 1.2–3.3) and 16% for fluconazole (OR 2.4, 95% CI 1.2–4.8).131
24.13.2 Fluconazole
Fluconazole is an azole antifungal. The azoles are synthetic antifungals with a triazole ring like fluconazole or an imidazole ring. They are fungistatic except at high concentrations and act by inhibiting a fungal cytochrome P450 enzyme involved in cell membrane ergosterol synthesis. Fluconazole can be given IV or orally, has excellent CSF penetration and is less nephrotoxic than amphotericin B, although can be hepatotoxic.132 Resistance may develop with prolonged or extensive use.132,133 Candida albicans species are usually sensitive, C. krusei is usually resistant to fluconazole and C. glabrata has reduced susceptibility, described as dose-dependent susceptibility (see Table 16.2).
A loading dose of 25 mg/kg is recommended134,135 and the treatment dose of fluconazole in neonatal fungal infection is 12 mg/kg/day IV or oral once daily.136
24.13.3 Echinocandins
The echinocandins are semi-synthetic cyclic lipopeptide antifungals which interfere with glucan formation and cause fungal cell lysis. They are active against most Candida species, Aspergillus and Pneumocystis, but inactive against zygomycetes and other less common moulds.137,138 However, C. parapsilosis has reduced susceptibility in vitro to echinocandins, although the clinical significance is unknown. Micafungin,139 anidulafungin 140 and caspofungin 141 have all been used in neonates.
A small randomized controlled trial (RCT) from Egypt compared caspofungin and amphotericin B for the treatment of invasive candidiasis.142 There was no significant difference in mortality, although the authors reported caspofungin as superior in terms of a ‘favourable response’, driven mainly by stopping amphotericin B in 29% of infants due to toxicity.142 There are insufficient data from good quality RCTs to justify echinocandin use in neonates except as salvage therapy.
24.13.4 Flucytosine (5-fluorocytosine, 5-flucytosine)
Flucytosine is a synthetic fluorinated pyrimidine antifungal which inhibits fungal DNA synthesis when converted to 5-fluorouracil. It is active against about 90% of C. albicans strains, against other Candida species except C. krusei and against Cryptococcus species but has little activity against other yeasts or moulds (see Table 16.2). Resistance commonly develops during monotherapy, so flucytosine should only be used in combination with another antifungal. It is well absorbed orally, achieves good CSF levels and is excreted renally, so has been used in combination therapy of renal candidiasis and of Candida meningitis. Although often used in combination with amphotericin B, there is no evidence from trials that adding flucytosine improves the outcome of Candida infections compared to monotherapy with amphotericin B or fluconazole. There is considerable individual variation in serum concentration and in renal clearance after oral dosing.143 Therapeutic drug monitoring (TDM) is important. The recommended dose is 25–100 mg/kg once daily, modified based on drug levels.143
24.14 Trimethoprim– sulfamethoxazole (cotrimoxazole)
Trimethoprim–sulfamethoxazole (TMP-SMX, cotrimoxazole) is a fixed ratio combination (1:5) of the synthetic diaminopyridine antibiotic trimethoprim and the sulphonamide antibiotic sulfamethoxazole. It is active against most community strains of MRSA, Acinetobacter species, B. cepacia, S. maltophilia, Pneumocystis, B. pertussis and some rapid growing mycobacteria (M. kansasii and M. fortuitum).
Although sulphonamides are generally contraindicated in the neonatal period because of the risk of causing kernicterus by displacing bilirubin bound to albumin and because of haematologic adverse effects, trimethoprim–sulfamethoxazole has been used occasionally in neonates because of its unique spectrum of activity. The greatest experience is with prophylactic trimethoprim–sulfamethoxazole for HIV-exposed infants: 6 months of prophylaxis is well tolerated, although treatment is not started until 4–6 weeks of age.144,145
There are very limited data on the use of trimethoprim–sulfamethoxazole in neonates. The only pharmacokinetic study recommended a loading dose of sulfamethoxazole 10 mg/kg with trimethoprim 3 mg/kg, and a maintenance dose of sulfamethoxazole 3 mg/kg with trimethoprim 1 mg/kg 12 hourly,146 which are not available (the fixed dose combination would allow a loading dose of sulfamethoxazole 10 mg/kg + trimethoprim 2 mg/kg and a maintenance dose of sulfamethoxazole 3 mg/kg with trimethoprim 0.6 mg/kg 12 hourly).
24.15 Chloramphenicol
Chloramphenicol is produced by soil streptomycetes although commercial chloramphenicol is synthesized. It inhibits bacterial protein synthesis by competing with messenger RNA for ribosomal binding sites. Chloramphenicol is active against a wide range of gram-positive, gram-negative, anaerobic and intracellular organisms. It is cheap and well absorbed so can be given orally or intravenously. Because of its excellent CSF penetration, it was used extensively to treat neonatal and infant meningitis.147 The major problems are severe toxicity (grey baby syndrome 148,149 and aplastic anaemia 150
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


