Otoniel Martínez-Maza
Jonathan S.Berek
Advances in molecular biology and genetics have improved our understanding of basic biologic concepts and disease development. The knowledge acquired with the completion of the human genome project, the data available through The Cancer Genome Atlas (TCGA), the development of novel diagnostic modalities, such as the microarray technology for the analysis of DNA and proteins, and the emergence of treatment strategies that target specific disease mechanisms all have an increasing impact on the specialty of obstetrics and gynecology.
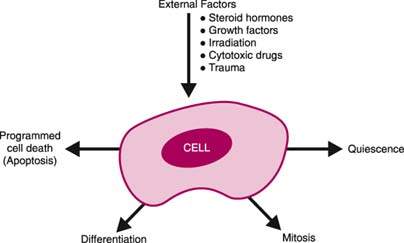
Normal cells are characterized by discrete metabolic, biochemical, and physiologic mechanisms. Specific cell types differ with respect to their mainly genetically determined responses to external influences (Fig. 6.1). An external stimulus is converted to an intracellular signal, for example, via a cell membrane receptor. The intracellular signal is transferred to the nucleus and generates certain genetic responses that lead to changes in cellular function, differentiation, and proliferation. Although specific cell types and tissues exhibit unique functions and responses, many basic aspects of cell biology and genetics are common to all eukaryotic cells.
Figure 6.1 External stimuli affect the cell, which has a specific coordinated response.
Cell Cycle
Normal Cell Cycle
Adult eukaryotic cells possess a well-balanced system of continuous production of DNA (transcription) and proteins (translation). Proteins are constantly degraded and replaced depending on the specific cellular requirements. Cells proceed through a sequence of phases called the cell cycle, during which the DNA is distributed to two daughter cells (mitosis) and subsequently duplicated (synthesis phase). This process is controlled at key checkpoints that monitor the status of a cell, for example, the amount of DNA present. The cell cycle is regulated by a small number of heterodimeric protein kinases that consist of a regulatory subunit (cyclin) and a catalytic subunit (cyclin-dependent kinase). Association of a cyclin with a cyclin-dependent kinase (CdkC) determines which proteins will be phosphorylated at a specific point during the cell cycle.
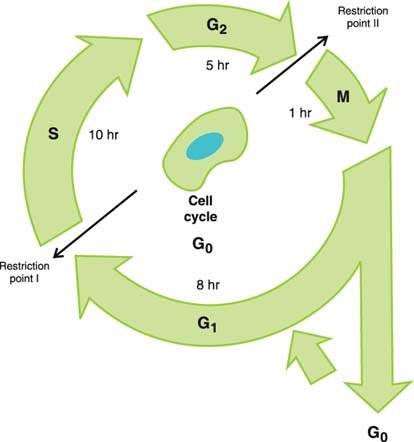
The cell cycle is divided into four major phases: M phase (mitosis), G1 phase (period between mitosis and initiation of DNA replication), S phase (DNA synthesis), and G2 phase (period between completion of DNA synthesis and mitosis) (Fig. 6.2). Postmitotic cells can “exit” the cell cycle into the so-called G0 phase and remain for days, weeks, or even a lifetime without further proliferation. The duration of the cell cycle may be highly variable, although most human cells complete the cell cycle within approximately 24 hours. During a typical cell cycle, mitosis lasts about 30 to 60 minutes, the G1 phase 7 to 10 hours, S phase 10 hours, and G2 phase 5 hours. With respect to the cell cycle, there are three subpopulations of cells:
Figure 6.2 The cell cycle.
Red blood cells, striated muscle cells, uterine smooth muscle cells, and nerve cells are terminally differentiated. Other cells, such as fibroblasts, exit from the G1 phase into the G0 phase and are considered to be out of the cell cycle. These cells enter the cell cycle following exposure to specific stimuli, such as growth factors and steroid hormones. Dividing cells are found in the gastrointestinal tract, the skin, and the cervix.
G1 Phase
In response to specific external stimuli, cells enter the cell cycle by moving from the G0 phase into the G1 phase. The processes during G1 phase lead to the synthesis of enzymes and regulatory proteins necessary for DNA synthesis during S phase and are mainly regulated by G1 cyclin-dependent kinase–cyclin complexes (G1CdkC). Complexes of G1CdkC induce degradation of the S phase inhibitors in late G1. Release of the S phase CdkC complex subsequently stimulates entry into the S phase. Variations in the duration of the G1 phase of the cell cycle, ranging from less than 8 hours to longer than 100 hours, account for the different generation times exhibited by different types of cells.
S Phase
The nuclear DNA content of the cell is duplicated during the S phase of the cell cycle. The S phase CdkC complex activates proteins of the DNA prereplication complexes that assemble on DNA replication origins during G1. The prereplication complex activates initiation of DNA replication and inhibits the assembly of new prereplication complexes. This inhibition ascertains that each chromosome is replicated only once during the S phase.
G2 Phase
RNA and protein synthesis occurs during the G2 phase of the cell cycle. The burst of biosynthetic activity provides the substrates and enzymes to meet the metabolic requirements of the two daughter cells. Another important event that occurs during the G2 phase of the cell cycle is the repair of errors of DNA replication that may have occurred during the S phase. Failure to detect and correct these genetic errors can result in a broad spectrum of adverse consequences for the organism and the individual cell (1). Defects in the DNA repair mechanism are associated with an increased incidence of cancer (2). Mitotic CdkC complexes are synthesized during the S and G2 phases, but are inactive until DNA synthesis is completed.
M Phase
Nuclear–chromosomal division occurs during the mitosis or M phase. During this phase, the cellular DNA is equally distributed to each of the daughter cells. Mitosis provides a diploid (2n) DNA complement to each somatic daughter cell. Following mitosis, eukaryotic mammalian cells contain diploid DNA, reflecting a karyotype that includes 44 somatic chromosomes and an XX or XY sex chromosome complement. Exceptions to the diploid cellular content include hepatocytes (4n) and the functional syncytium of the placenta.
Mitosis is divided into prophase, metaphase, anaphase, and telophase. Mitotic CdkC complexes induce chromosome condensation during the prophase, assembly of the mitotic spindle apparatus, and alignment of the chromosomes during the metaphase. Activation of the anaphase promoting complex (APC) leads to inactivation of the protein complexes that connect sister chromatids during metaphase, permitting the onset of anaphase. During anaphase, sister chromatids segregate to opposite spindle poles. The nuclear envelope breaks down into multiple small vesicles early in mitosis and reforms around the segregated chromosomes as they decondense during telophase. Cytokinesis is the process of division of the cytoplasm that segregates the endoplasmic reticulum and the Golgi apparatus during mitosis. After completion of mitosis, cells enter the G1 phase and either re-enter the cell cycle or remain in G0.
Ploidy
After meiosis, germ cells contain a haploid (1n) genetic complement. After fertilization, a 46,XX or 46,XY diploid DNA complement is restored. Restoration of the normal cellular DNA content is crucial to normal function. Abnormalities of cellular DNA content cause distinct phenotypic abnormalities, as exemplified by hydatidiform molar pregnancy (see Chapter 39). In a complete hydatidiform mole, an oocyte without any nuclear genetic material (e.g., an empty ovum) is fertilized by one sperm. The haploid genetic content of the fertilized ovum is then duplicated, and the diploid cellular DNA content is restored, resulting in a homozygous 46,XX gamete. Less often, a complete hydatidiform mole results from the fertilization of an empty ovum by two sperm, resulting in a heterozygous 46,XX or 46,XY gamete. In complete molar pregnancies, the nuclear DNA is usually paternally derived, embryonic structures do not develop, and trophoblast hyperplasia occurs. Rarely, complete moles are biparental. This karyotype seems to be found in patients with recurrent hydatiform moles and is associated with a higher risk of persistent trophoblastic disease.
A partial hydatiform mole follows the fertilization of a haploid ovum by two sperm, resulting in a 69,XXX, 69,XXY, or 69,XYY karyotype. A partial mole contains paternal and maternal DNA, and both embryonic and placental development occur. Both the 69,YYY karyotype and the 46,YY karyotype are incompatible with embryonic and placental development. These observations demonstrate the importance of maternal genetic material, in particular the X chromosome, in normal embryonic and placental development.
In addition to total cellular DNA content, the chromosome number is an important determinant of cellular function. Abnormalities of chromosome number, which often are caused by nondisjunction during meiosis, result in well-characterized clinical syndromes such as trisomy 21 (Down syndrome), trisomy 18, and trisomy 13.
Genetic Control of the Cell Cycle
Cellular proliferation must occur to balance normal cell loss and maintain tissue and organ integrity. This process requires the coordinated expression of many genes at discrete times during the cell cycle (3). In the absence of growth factors, cultured mammalian cells are arrested in the G0 phase. With the addition of growth factors, these quiescent cells pass through the so-called restriction point 14 to 16 hours later and enter the S phase 6 to 8 hours thereafter. The restriction point or G1/S boundary marks the point at which a cell commits to proliferation. A second checkpoint is the G2/M boundary, which marks the point at which repair of any DNA damage must be completed (4–7). To successfully complete the cell cycle, a number of cell division cycle (cdc) genes are activated.
Cell Division Cycle Genes
Among the factors that regulate the cell cycle checkpoints, proteins encoded by the cdc2 family of genes and the cyclin proteins appear to play particularly important roles (8,9). Growth factor–stimulated mammalian cells express early-response or delayed-response genes, depending on the chronological sequence of the appearance of specific RNAs. The early- and delayed-response genes act as nuclear transcription factors and stimulate the expression of a cascade of other genes. Early-response genes such as c-Jun and c-Fos enhance the transcription of delayed-response genes such as E2Fs. E2F transcription factors are required for the expression of various cell cycle genes and are functionally regulated by the retinoblastoma (Rb) protein. Binding of Rb to E2F converts E2F from a transcriptional activator to a repressor of transcription. Phosphorylation of Rb inhibits its repressing function and permits E2F-mediated activation of genes required for entry into the S phase. Cdk4-cyclin D, Cdk6-cyclin D, and Cdk2-cyclin E complexes cause phosphorylation of Rb, which remains phosphorylated throughout the S, G2, and M phases of the cell cycle. After completion of mitosis, a decline of the level of Cdk-cyclins leads to dephosphorylation of Rb by phosphatases and, consequently, an inhibition of E2F in the early G1 phase.
Cdks are being evaluated as targets for cancer treatments because they are frequently overactive in cancer disease and Cdk-inhibiting proteins are dysfunctional. The Cdk4 inhibitor P1446A-05, for example, specifically inhibits Cdk4-mediated G1-S phase transition, arresting cell cycling and inhibiting cancer cell growth (10). SNS-032 selectively binds to Cdk2, -7, and -9, preventing their phosphorylation and activation and subsequently preventing cell proliferation.
As cells approach the G1-S phase transition, synthesis of cyclin A is initiated. The Cdk2-cyclin A complex can trigger initiation of DNA synthesis by supporting the prereplication complex. The p34 cdc2 protein and specific cyclins form a complex heterodimer referred to as mitosis-promoting factor (MPF), which catalyzes protein phosphorylation and drives the cell into mitosis. Cdk1 assembles with cyclin A and cyclin B in the G2 phase and promotes the activity of the MPF. Mitosis is initiated by activation of the cdc gene at the G2-M checkpoint (11,12). Once the G2-M checkpoint is passed, the cell undergoes mitosis. In the presence of abnormally replicated chromosomes, progression past the G2-M checkpoint does not occur.
The p53 tumor suppressor gene participates in cell cycle control. Cells exposed to radiation therapy exhibit an S-phase arrest that is accompanied by increased expression of p53. This delay permits the repair of radiation-induced DNA damage. In the presence of p53 mutations, the S-phase arrest that normally follows radiation therapy does not occur (13,14). The wild type p53 gene can be inactivated by the human papillomavirus E6 protein, preventing S-phase arrest in response to DNA damage (15).
Apoptosis
The regulation and maintenance of normal tissue requires a balance between cell proliferation and programmed cell death, or apoptosis. When proliferation exceeds programmed cell death, the result is hyperplasia. When programmed cell death exceeds proliferation, the result is atrophy. Programmed cell death is a crucial concomitant of normal embryologic development. This mechanism accounts for deletion of the interdigital webs, palatal fusion, and development of the intestinal mucosa (16–18). Programmed cell death is also an important phenomenon in normal physiology (19). The reduction in the number of endometrial cells following alterations in steroid hormone levels during the menstrual cycle is, in part, a consequence of programmed cell death (20,21). In response to androgens, granulosa cells undergo programmed cell death (e.g., follicular atresia) (22).
Programmed cell death, or apoptosis, is an energy-dependent, active process that is initiated by the expression of specific genes. This process is distinct from cell necrosis, although both mechanisms result in a reduction in total cell number. In programmed cell death, cells shrink and undergo phagocytosis. Conversely, groups of cells expand and lyse when undergoing cell necrosis. The process is energy independent and results from noxious stimuli. Programmed cell death is triggered by a variety of factors, including intracellular signals and exogenous stimuli such as radiation exposure, chemotherapy, and hormones. Cells undergoing programmed cell death may be identified on the basis of histologic, biochemical, and molecular biologic changes. Histologically, apoptotic cells exhibit cellular condensation and fragmentation of the nucleus. Biochemical correlates of impending programmed cell death include an increase in transglutaminase expression and fluxes in intracellular calcium concentration (23).
Programmed cell death emerged as an important factor in the growth of neoplasms. Historically, neoplastic growth was characterized by uncontrolled cellular proliferation that resulted in a progressive increase in tumor burden. It is recognized that the increase in tumor burden associated with progressive disease reflects an imbalance between cell proliferation and cell death. Cancer cells fail to respond to the normal signals to stop proliferating, and they may fail to recognize the physiologic signals that trigger programmed cell death.
Modulation of Cell Growth and Function
The normal cell exhibits an orchestrated response to the changing extracellular environment. The three groups of substances that signal these extracellular changes are steroid hormones, growth factors, and cytokines. The capability to respond to these stimuli requires a cell surface recognition system, intracellular signal transduction, and nuclear responses for the expression of specific genes in a coordinated fashion. Among the genes that participate in control of cell growth and function, proto-oncogenes and tumor suppressor genes are particularly important. More than 100 proto-oncogene products that contribute to growth regulation have been identified (24) (Table 6.1). As a group, proto-oncogenes exert positive effects upon cellular proliferation. In contrast, tumor suppressor genes exert inhibitory regulatory effects on cellular proliferation (Table 6.2).
Steroid Hormones
Steroid hormones play a crucial role in reproductive biology and in general physiology. Among the various functions, steroid hormones influence pregnancy, cardiovascular function, bone metabolism, and an individual’s sense of well-being. The action of steroid hormones is mediated via extracellular signals to the nucleus to affect a physiologic response.
Table 6.1 Proto-Oncogenes
| Proto-oncogenes | Gene Product/Function |
| Growth factors | |
| Fibroblast growth factor | |
| fgf-5 | |
| Sis | Platelet-derived growth factor beta |
| hst, int-2 | |
| Transmembrane receptors | |
| erb-B | Epidermal growth factor (EGF) receptor |
| HER-2/neu | EGF-related receptor |
| Fms | Colony-stimulating factor (CSF) receptor |
| Kit | Stem cell receptor |
| Trk | Nerve growth factor receptor |
| Inner-membrane receptor | |
| bcl-2 | |
| Ha-ras, N-ras, N-ras | |
| fgr, lck, src, yes | |
| Cytoplasmic messengers | |
| Crk | |
| cot, plm-1, mos, raf/mil | |
| Nuclear DNA binding proteins | |
| erb-B1 | |
| jun, ets-1, ets-2,fos, gil 1,rel, ski, vav | |
| lyl-1, maf, myb, myc, L- myc, N-myc, evi-1 |
Table 6.2 Tumor Suppressor Genes
| p53 | Mutated in as many as 50% of solid tumors |
| Rb | Deletions and mutations predispose to retinoblastoma |
| PTEN | Dual specificity phosphatase that represses PI3-kinase/Akt pathway activation with negative effect on cell growth |
| P16INK4a | Binds to cylin-CDK4 complex inhibiting cell cycle progression |
| FHIT | Fragile histidine triad gene with tumor suppressor function via unknown mechanisms |
| WT1 | Mutations are correlated with Wilms’ tumor |
| NF1 | Neurofibromatosis gene |
| APC | Associated with colon cancer development in patients with familial adenomatous |
Estrogens exert a variety of effects on growth and development of different tissues. The effects of estrogens are mediated via estrogen receptors (ER), intracellular proteins that function as ligand-activated transcription factors and belong to the nuclear receptor superfamily (25). Two mammalian ERs have been identified: ERα and ERβ. The structure of both receptors is similar and consists of six domains named A through F from the N- to C-terminus, encoded by 8 to 9 exons (26). Domains A and B are located at the N-terminus and contain an agonist-independent transcriptional activation domain (activation function 1, or AF-1). The C domain is a highly conserved central DNA-binding domain composed of two zinc fingers through which ER interacts with the major groove and the phosphate backbone of the DNA helix. The C-terminus of the protein contains domains E and F and functions as ligand-binding domain (LBD–domain E) and AF-2–domain F (Fig. 6.3).
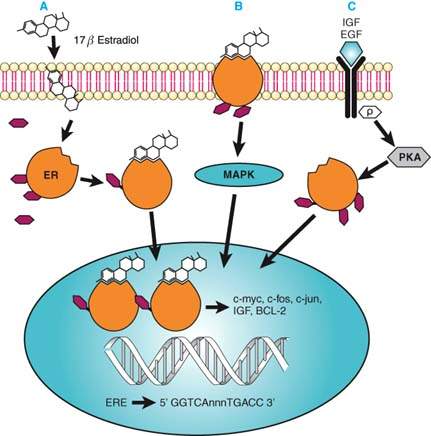
Activation of transcription via the ER is a multistep process. The initial step requires activation of the ER via various mechanisms (Fig. 6.4). For example, estrogens such as 17β-estradiol can diffuse into the cell and bind to the LBD of the ER. Upon ligand binding, the ER undergoes conformational changes followed by a dissociation of various bound proteins, mainly heat shock proteins 90 and 70 (Hsp90 and Hsp70). Activation of the ER also requires phosphorylation by several protein kinases, including casein kinase II, protein kinase A, and components of the Ras/Mapk (mitogen-activated protein kinase) pathway (26). Four phosphorylation sites of the ER are clustered in the NH2 terminus with the AF-1 region.
The activated ER elicits a number of different genomic as well as nongenomic effects on intracellular signaling pathways. The classical steroid signaling pathway involves binding of the activated estrogen receptor to an estrogen responsive element (ERE) on the genome as homodimers and subsequent stimulation of transcription (27,28). The minimal consensus sequence for the ERE is a 13 bp palindromic inverted repeat (IR) and is defined as 5′-GGTCAnnnTGACC-3′. Genes that are regulated by activated ERs include early gene responses such as c-myc, c-fos, and d-jun, and genes encoding for growth factors such as insulin growth factor (IGF-1 and IGF-2), epidermal growth factor (EGF), transforming growth factor-α, and colony-stimulating factor (CSF-1).
Figure 6.3 Structure of the two mammalian estrogen receptors. ERα (595 amino acids) and ERβ (530 amino acids) consist of six domains (A–F from the N- to C-terminus). Domains A and B at the N-terminus contain an agonist-independent transcriptional activation domain (activation function 1, or AF-1). The C domain is the central DNA-binding sequence (DBD). Domains E and F function as ligand binding domain (LBD) and activation function 2 (AF-2). Also shown is the structure of the ER ligand 17β-estradiol.
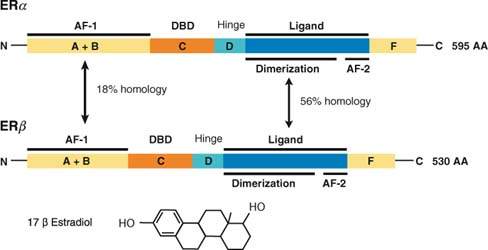
Figure 6.4 Activation of estrogen receptor mediated transcription. Intracellular estrogen receptor signaling is mediated via different pathways. A: 17β-estrodiol diffuses through the cell membrane and binds to cytoplasmic ER. The ER is subsequently phosphorylated, undergoes dimerization, and binds to the estrogen response element (ERE) on the promoter of an estrogen responsive gene. B: Estrogen ligand binds to membrane-bound ER and activates the mitogen-activated protein kinase (MAPK) pathways that support ER-mediated transcription. C: Binding of cytokines such as insulinlike growth factor (IGF) or epidermal growth factor to their membrane receptor can cause activation of protein kinases like PKA, which subsequently activates ER by phosphorylation.
In addition to the described genomic effects of estrogens, there is growing evidence for nongenomic effects of estrogens on intracellular signal transduction pathways. These effects include rapid activation of the adenylate cyclase, which results in cyclic adenosine monophosphate (cAMP)–dependent activation of protein kinase A (PKA) (29). Estrogens can stimulate the MAPK pathway and rapidly activate the Erk1/Erk2 proteins.
Various ligands with different affinities to the ER were developed and are called selective estrogen receptor modulators (SERMs). Tamoxifen, for example, is a mixed agonist/ antagonist for ERα, but it is a pure antagonist for ERβ. The ERβ receptor is ubiquitously expressed in hormone-responsive tissues, whereas the expression of ERα fluctuates in response to the hormonal milieu. The cellular and tissue effects of an estrogenic compound appear to reflect a dynamic interplay between the actions of these estrogen receptor isoforms. These observations underscore the complexity of estrogen interactions with both normal and neoplastic tissue. Mutations of hormone receptors and their functional consequences illustrate their important contributions to normal physiology. For example, absence of ERα in a male human was reported (30). The clinical sequelae attributed to this mutation include incomplete epiphyseal closure, increased bone turnover, tall stature, and impaired glucose tolerance. The androgen insensitivity syndrome is caused by mutations of the androgen receptor (31). Mutations of the receptors for growth hormone and thyroid-stimulating hormone result in a spectrum of phenotypic alterations. Mutations of hormone receptors may also contribute to the progression of neoplastic disease and resistance to hormone therapy (32,33).
Growth Factors
Growth factors are polypeptides that are produced by a variety of cell types and exhibit a wide range of overlapping biochemical actions. Growth factors bind to high-affinity cell membrane receptors and trigger complex positive and negative signaling pathways that regulate cell proliferation and differentiation (34). In general, growth factors exert positive or negative effects upon the cell cycle by influencing gene expression related to events that occur at the G1-S cell cycle boundary (35).
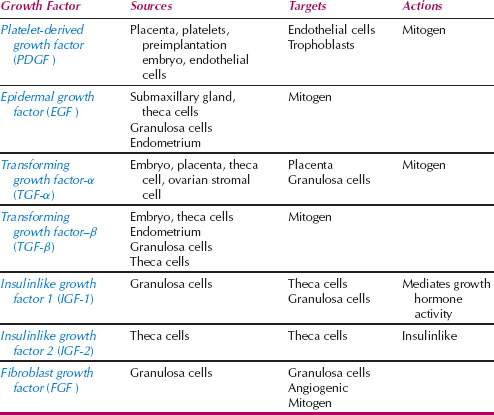
Because of their short half-life in the extracellular space, growth factors act over limited distances through autocrine or paracrine mechanisms. In the autocrine loop, the growth factor acts on the cell that produced it. The paracrine mechanism of growth control involves the effect of growth factors on another cell in proximity. Growth factors that play an important role in female reproductive physiology are listed in Table 6.3. The biologic response of a cell to a specific growth factor depends on a variety of factors, including the cell type, the cellular microenvironment, and the cell cycle status.
Table 6.3 Growth Factors that Play Important Roles in Female Reproductive Physiology
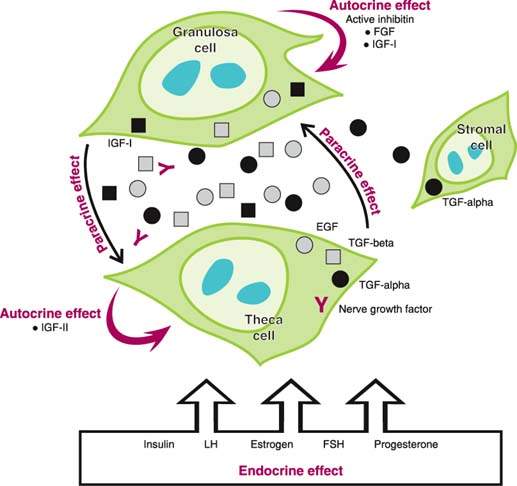
The regulation of ovarian function occurs through autocrine, paracrine, and endocrine mechanisms (36–42). The growth and differentiation of ovarian cells are particularly influenced by the insulinlike growth factors (IGF) (Fig. 6.5). IGFs amplify the actions of gonadotropin hormones on autocrine and paracrine growth factors found in the ovary. IGF-1 acts on granulosa cells to cause an increase in cAMP, progesterone, oxytocin, proteoglycans, and inhibin. On theca cells, IGF-1 causes an increase in androgen production. Theca cells produce tumor necrosis factor-α (TNF-α) and EGF, which are regulated by follicle-stimulating hormone (FSH). Epidermal growth factor acts on granulosa cells to stimulate mitogenesis. Follicular fluid contains IGF-1, IGF-2, TNF-α, TNF-β, and EGF. Disruption of these autocrine and paracrine intraovarian pathways may be the basis of polycystic ovarian disease, disorders of ovulation, and ovarian neoplastic disease.
Figure 6.5 The regulation of ovarian function occurs through autocrine, paracrine, and endocrine mechanisms.
Transforming growth factor-β (TGF-β) activates intracytoplasmic serine threonine kinases and inhibits cells in the late G1 phase of the cell cycle (42). It appears to play an important role in embryonic remodeling. Mullerian-inhibiting substance (MIS), which is responsible for regression of the mullerian duct, is structurally and functionally related to TGF-β (43). TGF-α is an EGF homologue that binds to the EGF receptor and acts as an autocrine factor in normal cells. As with EGF, TGF-α promotes entry of G0 cells into the G1 phase of the cell cycle. The role of growth factors in endometrial growth and function was the subject of several reviews (37–42). Similar to the ovary, autocrine, paracrine, and endocrine mechanisms of control also occur in endometrial tissue.
Intracellular Signal Transduction
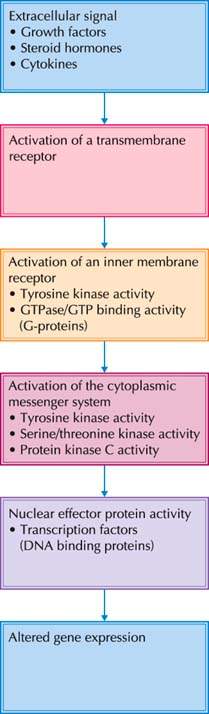
Growth factors trigger intracellular biochemical signals by binding to cell membrane receptors. In general, these membrane-bound receptors are protein kinases that convert an extracellular signal into an intracellular signal. The interaction between growth factor ligand and its receptor results in receptor dimerization, autophosphorylation, and tyrosine kinase activation. Activated receptors in turn phosphorylate substrates in the cytoplasm and trigger the intracellular signal transduction system (Fig. 6.6). The intracellular signal transduction system relies on serine threonine kinases, src-related kinases, and G proteins. Intracellular signals activate nuclear factors that regulate gene expression. Many of the proteins that participate in the intracellular signal transduction system are encoded by proto-oncogenes that are divided into subgroups based on their cellular location or enzymatic function (44,45) (Fig. 6.7).
Figure 6.6 Pathways of intracellular signal transduction.