(1)
Section of Pharmacology, Department of Medicine, University of Perugia, Building D, 2nd floor, Severi Square 1, San Sisto, Perugia, I-06132, Italy
Introduction
Owing to their powerful anti-inflammatory and immunomodulatory actions, glucocorticoids (GCs) are widely used to treat both acute and chronic inflammatory conditions. GCs also induce immunosuppression and therefore are administered after organ transplantation, during severe allergic reactions or autoimmune flare-ups, and as part of chemotherapy regimens. However, long-term and intense stimulation of GC receptors (GRs) causes adverse effects that are collectively termed Cushing’s syndrome.
During the past century, researchers have produced more potent GCs that have longer half-lives and lack virtually all of the mineral corticoid effects (e.g., betamethasone and dexamethasone) compared with endogenous GC (cortisol) and the first synthetic GC (prednisolone).
Molecules that have entered the clinic in the past 20 years are GC analogues that decrease the risk-to-benefit ratios for the long-term topical treatment of some diseases. These molecules have improved pharmacokinetic properties and have increased the desired effects in peripheral tissues while simultaneously decreasing the unwanted systemic effects. However, it seems they have a mechanism of action identical to that of older GCs. In this chapter, we present the molecular and cellular mechanisms responsible for the powerful anti-inflammatory and immunosuppressive effects of GCs.
Molecular Mechanisms of Glucocorticoids
GCs exert different effects in different tissues. For example, GCs can trigger apoptosis in some lymphocytes but protect against cell death in other lymphocytes or parenchymal cells in inflamed tissues. Array studies evaluating the messenger RNA (mRNA) levels from a diverse cell population demonstrated that these differences are because the majority of genes modulated by GCs in a certain cell type are not expressed in cells with other phenotypes. In the following paragraphs, we explain how it is possible that a drug that targets just one receptor causes this amazing variety by both genomic and nongenomic effects, as summarized in Fig. 1.
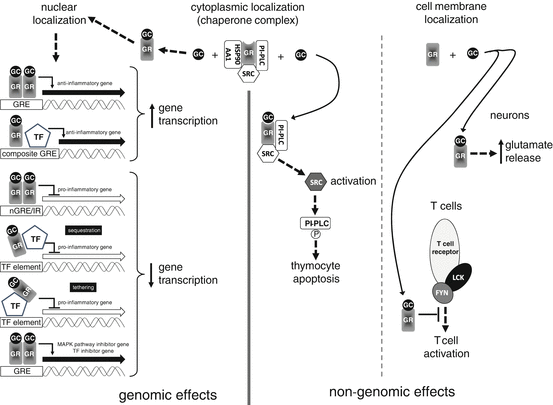
Fig. 1
Mechanism of action of glucocorticoids. Glucocorticoids (GCs) act through two mechanisms: genomic (main mechanism) and nongenomic (accessory but immediately effective). GCs pass from the circulation into cells where they bind GC receptor (GR), located in a cytoplasmic chaperone complex (middle), in complexes located in the cell membranes (right) or in other cellular structures (not shown). The chaperone complex is formed by several proteins including heat shock protein 90 kDa alpha, class A member 1 (HSP90AA1), phosphatidylinositol-specific phospholipase C (PI-PLC) and SRC proto-oncogene, nonreceptor tyrosine kinase (SRC). Following nuclear translocation of activated GR (left), genomic effects are exerted by at least six mechanisms: (1) upregulation of gene transcription by binding GC-response elements (GRE); (2) upregulation of gene transcription by activation of transcription factors (TF) that bind DNA next to a GR-binding element (composite GRE); (3) downregulation of gene transcription by binding negative GRE (nGRE) characterized by inverted repeat (IR); (4) downregulation of gene transcription by binding TFs and inhibiting their binding to DNA (sequestration); (5) downregulation of gene transcription by binding TFs and inhibiting their transcriptional activity (tethering); (6) upregulation of gene transcription by binding GRE in the promoter region of genes coding for inhibitors of TF (conclusively leading to downregulation of gene transcription) or proinflammatory pathways, such as the MAPK pathway. Nongenomic effects depend on the localization of GR and the GR-bound proteins. In thymocytes, activated GR activates SRC that, in turn, phosphorylates PI-PLC, leading to thymocyte apoptosis. In neurons of the prefrontal and frontal cortex, GR is present is synaptosomes and in presynaptic spines and its activation increases the readily releasable pool of glutamate vesicles. In T cells, GR is close to T cell receptor (TCR) complex and GR activation inhibits TCR-dependent activation of lymphocyte-specific protein tyrosine kinase (LCK) and FYN oncogene related to SRC, FGR, and YES (FYN), inhibiting T cell activation
Genomic Effects
Owing to their hydrophobic nature, GCs pass from the circulation into cells where they bind the ubiquitously expressed GC receptor (GR). GR is located in the cytoplasm where it exists in a multimeric chaperone complex composed of heat shock proteins (HSP90AA1 and HSP70), HSP-binding phosphoprotein p23, immunophilins (FKBP51, FKBP52, Cyp44, and PP5), Hip, Hop, and other factors [76, 100]. The chaperone heat shock protein 90 kDa alpha, class A member 1 (HSP90AA1), maintains GR in a favorable conformational state that is required for high-affinity ligand binding and cytoplasmic retention.
There are two main isoforms of human GR (hGR), the predominant hGRα and hGRβ. Each of the GR mRNA species, α and β, produces at least eight functional GR N-terminal isoforms via translational modifications, each with potentially unique transcriptional activities [79]. For example, the GRα isoforms, GR-A, GR-B, and GR-C, induce Jurkat cell apoptosis, whereas the GR-D isoform does not [103]. Moreover, GRs can be phosphorylated and sumoylated, thereby modulating their function and half-life [15, 79]. hGRβ is unable to bind any GCs and is transcriptionally inactive; thus, it acts as a dominant-negative regulator of hGRα [69].
Upon GC binding, GRα changes conformation and owing to the chaperone machinery, the receptor translocates to the nucleus [100] to positively and negatively regulate gene transcription.
Upregulation of Gene Transcription
The primary manner by which activated GRα (herein referred to as GR) upregulates gene transcription (also known as transactivation) is via dimerization and binding to GC-response elements (GREs) that are present in one or more copies in the promoter regions of hundreds of genes. In some cases, the GRE is far from the transcriptional start site. Although the canonical GRE was defined as the palindromic sequence A-G-A-A-C-A-N-N-N-T-G-T-T-C-T (where N indicates any nucleotide), many variations are possible, particularly in the 3′ half site. The variations are described by the sequence N-G-N-(A,T)-C-(A,G,T)-N-N-N-(A,T)-G-T-(C,T)-C-T [98]. The interaction between GR and DNA induces a variety of coregulatory factors – including Brg1, histone acetyl transferases such as CBP or SRC-1, and RNA polymerase II – to physically associate with GR, be recruited to chromatin, and ultimately drive transcription [17]. In this context, the cellular effects of GR also depend on the types and expression levels of coactivators.
In the promoters of some genes, GR monomers or dimers collaborate with other transcription factors to cooperatively enhance transcription. This can result from GR and other transcription factors binding to adjacent binding sites (composite elements) or from GR binding DNA-bound transcription factors (tethering) [60, 89]. The gene transcription effects occur in chromatin where the DNA is accessible independent of hormone action [46], thus modulating distinct genomic loci in different cells and resulting in tissue- and cell-specific effects.
Several, but not all, metabolic effects of GC depend on GR homodimerization and GRE binding. The same mechanism is responsible for the increased production of several genes with anti-inflammatory or immunosuppressive effects coding for inhibitors of proinflammatory cytoplasmic pathways (section “GC-activated anti-inflammatory and immunosuppressive pathways”), modulators of transcription factors (section “Downregulation of gene transcription by GR-dependent upregulation of inhibitory factors”), and other proteins such as tristetraprolin (TTP), a protein favoring destabilization and degradation of mRNAs coding for proinflammatory factors [86].
Downregulation of Gene Transcription
GR predominantly downregulates gene transcription of cytokines and other factors that contribute to the development of inflammation and immune response by inhibiting protein-to-protein interactions (also known as sequestration). This signaling mechanism is best characterized for the inhibition of transcription factors nuclear factor κB (NF-κB), activator protein-1 (AP-1), nuclear factor of activated T-cells and Smad3 [31]. For example, NF-κB is maintained in an inactive state via interaction with its inhibitor IκB. NF-κB is activated by proinflammatory stimuli, which causes IκB to be phosphorylated and dissociate from the IκB/NF-κB complex. GR can repress NF-κB-mediated gene activation by physically interacting with the p65 subunit of NF-κB, thereby sequestering p65 [58].
GR can also inhibit transcription factors such as NF-κB by forming a complex when they are bound to DNA and preventing the recruitment of transcriptional machinery (effect also known as tethering) [14]. The sequestration mechanism of GR action affects the activity of transcription factors expressed in the cell, causing tissue- and cell-specific effects.
In the promoter of genes that are downregulated by GC, the sequence C-T-C-C-(N)0–2-G-G-A-G-A has been described [92]. This sequence is suggested to represent a DNA-binding site called simple negative GRE (nGRE) or inverted repeat (IR) to which the GR homodimer binds. nGREs mediate transrepression by direct binding of activated GR that assembles a repressing complex. In other genes, nGREs are located close to DNA-binding sites for other transcription factors. Thus, GR inhibits transcription factor activity by competing with and displacing transcription factors from DNA [36]. For example, GR binding to the nGRE in the promoter of hFas ligand prevents NF-κB binding and inhibits Fas ligand expression [67]. In other cases, GR binding to the GRE or nGRE inhibits transcription factor binding to a DNA-binding site proximal to the GR binding site (so-called composite elements) [14].
Downregulation of Gene Transcription by GR-Dependent Upregulation of Inhibitory Factors
One mechanism that explains the powerful anti-inflammatory effects of GC is the very rapid, GRE-dependent upregulation of proteins that inhibit transcription factors. Among these proteins, glucocorticoid-induced leucine zipper (GILZ), IκB (an inhibitor of NF-κB) [5], and Krüppel-like factor 2 (KLF2; an inhibitor of NF-κB and AP-1) [30] are the best characterized.
GILZ was originally identified in 1997 during a systematic study of genes transcriptionally induced by GC [28]. It is one of the few genes induced by GC in nearly all immune cell types. The finding that GILZ silencing abrogates DEX antiproliferative activity [9] and reduces GC-mediated inhibition of cytokine-induced cyclooxygenase (COX)-2 expression [104] distinguished GILZ as a critical mediator of GC effects. GILZ overexpression modulates several cellular pathways [8]; however, it primarily inhibits transcription factors such as NF-κB and AP-1 [7, 34, 35, 61].
In addition to the GC-dependent effects on transcription factor modulators, which seem to be very rapid (of the order of hours), GR-dependent direct and indirect modulation of several transcription factors causes a striking amplification effect. This results in the modulation of thousands of genes in the subsequent days following GC treatment [22].
Nongenomic Effects
Although most GC immunosuppressive and anti-inflammatory effects are mediated through genomic effects that require hours to days, others take place within minutes (e.g., rapid cardiovascular protective effects, rapid clinical improvement of anaphylactic reactions, and effects on glutamate and GABA release). These cannot be explained by genomic mechanisms. Several hypotheses have been suggested to explain these effects, including aspecific effects on cell membrane fluidity or activation of G protein-coupled receptors specific for GCs [53, 93]. Although it is increasingly recognized that receptors activated by GCs are present in the plasma membrane and are capable of activating cytoplasmic pathways, a transmembrane receptor specific for GCs has not yet been described. By contrast, recent studies suggest that GC-free GRs are not confined to the GR chaperone complex but rather are present in other cellular locations, including the cell membrane [70], likely in association with other receptors or signaling molecules. In this case, GRα activation by GC promotes not only the activation of GRα but also of the other receptors/signaling molecules [90].
Indeed, several studies suggest that the nongenomic effects of GR depend on the particular localization of GR in cells. For example, in adenocarcinoma cells, GR binds proto-oncogene, nonreceptor tyrosine kinase (SRC) and activated GR releases SRC, which subsequently phosphorylates annexin-1. Phosphorylated annexin-1, in turn, displaces the adaptor protein Grb2 from epidermal growth factor receptor. Consequently, GR activation inhibits epidermal growth factor receptor-dependent activation of PLA2 and arachidonic acid release [27]. In thymocytes, GR binds phosphatidylinositol-specific phospholipase C and SRC, and activated GR determines the rapid phosphorylation and activation of phosphatidylinositol-specific phospholipase C due to SRC, which ultimately leads to thymocyte apoptosis [55]. In T cells, GR has a close physical and functional interaction with the T cell receptor (TCR) complex, and GR activation causes a rapid dissolution of TCR-linked GR complexes and inhibits Lck and Fyn, kinases immediately downstream of the TCR [52]. In neurons of the prefrontal and frontal cortex, GR is present in synaptosomes, as well as in presynaptic membranes and postsynaptic spines, and its activation increases the readily releasable pool of glutamate vesicles in synaptic terminals [95].
When considering the effects of GC on the central nervous system, it should be noted that neurosteroids (e.g., allopregnanolone and tetrahydrodeoxycorticosterone) target GABAA receptors, thereby potentiating GABAA receptor-mediated currents [88]. Although it has been shown that neurosteroid levels depend on local de novo synthesis and progesterone metabolism, the potential effects of GCs on neurosteroid production cannot be ruled out.
In conclusion, the nongenomic effects of GCs result from the binding of GRs located in the cell membrane (or other cellular structures) and the consequent activation of cytoplasmic pathways not involved in gene transcription.
GC-Activated Anti-inflammatory and Immunosuppressive Pathways
Virtually all cells of the adaptive and innate immune systems are modulated by GCs. The various mechanisms include inhibiting production of a large number of proinflammatory factors that are crucial for the development, maintenance, and outcome of inflammation and autoimmune diseases. This is the primary reason why GCs remain the most potent immunosuppressive drugs available. However, GCs modulate the functions of other cells and tissues, thus causing adverse effects during long-term treatment.
Although we will focus on effects of GCs at therapeutic doses, it is interesting to note that GCs at low concentrations, particularly endogenous GCs, favor maturation and differentiation of the immune system and other tissues, including muscles and the central nervous system. Thus, it is possible that GCs determine opposing effects (e.g., apoptosis and protection against apoptosis) [16] depending on their concentration as well as the functional status of the cell.
The anti-inflammatory and immunosuppressive effects of GCs have been attributed to their ability to inhibit the activity of the transcription factors, including NF-κB, AP-1, CREB, NF-AT, STAT, IRF3, and T-bet. These transcription factors are involved in the expression of many proinflammatory genes, including the cytokines interleukin (IL)-6, IL-1β, and tumor necrosis factor-α, the chemokines macrophage inflammatory protein and RANTES, the enzymes inducible nitric oxide synthase and COX-2, and the adhesion molecules intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin [21, 32, 50, 101].
GC-induced upregulation of anti-inflammatory genes plays a key role in their anti-inflammatory and immunosuppressive activity via several mechanisms including: (1) mitogen-activated protein kinase (MAPK) pathway inhibition (upregulation of dual-specificity phosphatase-1, docking protein-1, and GILZ); (2) prostaglandin synthesis inhibition [upregulation of annexin-1 and Clara cell 10 kDa (CC10)]; (3) inhibition of T cell activation [upregulation of Src-like adaptor protein (SLAP), IL-10, and IL-1 receptor antagonist], and (4) activation of regulatory T cells [upregulation of Forkhead box P3 (FoxP3) and IL-10].
Some of the upregulated genes have multiple concurrent functions. For example, GILZ binds and inhibits NF-κB and AP-1, as well as Ras and Raf, resulting in inhibition of both the Akt and ERK pathways, two MAPK pathways that contribute to the regulation of proinflammatory transcription factors and perpetuate inflammatory cascade activation [8–10]. GC-induced annexin-1 upregulation participates in the inhibition of phospholipase A2, an enzyme that initiates the cascade leading to prostaglandin production, inhibition of tumor necrosis factor-α upregulation, and induction of IL-10, an anti-inflammatory cytokine [38, 87]. Moreover, annexin-1 inhibits MAPK signaling, either directly by regulating components of the signaling cascade or indirectly by modulating other GC-induced proteins, such as GILZ [6].
Another mechanism by which GCs regulate inflammatory processes is through modulation of the expression and activity of a variety of kinases and phosphatases [14]. For example, GC upregulates expression of dual-specificity phosphatase-1, which is a negative feedback regulator of MAPK signaling [84].
GCs activate various pathways to inhibit cell growth and induce apoptosis. Thymocytes, particularly CD4+CD8+ double-positive thymocytes, are considered the prototype of GC-induced apoptosis because of their high sensitivity to GCs. In the past, several groups (including ours) attempted to find the crucial proapoptotic event(s) – including increased or decreased gene expression – that are definitively responsible for GC-induced apoptosis. Array studies indicate that several genes that participate in different cellular functions are modulated by GC treatment, causing an imbalance between pro- and antiapoptotic genes and thus inducing apoptosis (termed the network hypothesis) [16, 83]. The dysregulated genes include genes that control the redox status and the mitochondrial machinery (including Bcl-2, Bcl-X, and Bim), as well as the Tis11 family, which is involved in mRNA stability, and the ceramide pathway [16, 71]. Other genomic and nongenomic mechanisms participate in the induction of thymocyte apoptosis, such as Src kinase activation, GILZ upregulation, or plasma membrane potential modulation, and involve aSMase, caspases-3, -8, and -9, proteasomal degradation, and lysosomal pathways [26, 54, 55]. Even mature T cells can undergo GC-mediated apoptosis [105]. Apoptosis of leukemic cells is mediated by both genomic and nongenomic mechanisms. The nongenomic mechanism involves translocation of GRs to the mitochondria upon GC binding, thereby modifying their transmembrane potential [85].
Other GC-modulated genes inhibit apoptosis, such as inhibition of activation-induced cell death (AICD) in mature T cells, whose GC-dependent reduction of CD95L transcription is one of the involved mechanisms [13]. The antiapoptotic effects of GCs are observed in several parenchymal cell types, including endometrial cells and neurons, saving peripheral tissues and contributing to the anti-inflammatory effects of GCs [45, 72].
Effects on T Lymphocytes
Recent evidence suggests that both thymic regulatory T cells (tTregs) and peripheral regulatory T cells (pTreg) are needed to establish and maintain tolerance [42]. In autoimmune diseases, several reports have demonstrated decreased numbers of Tregs or defects in Treg suppressive activity, which is either intrinsic or due to partial resistance of activated effector T cells [40, 48, 94]. Moreover, Treg expansion has been described in patients with a milder autoimmune disease as compared with patients with more aggressive disease [2, 66]. T helper (Th) 1 cells are the predominant subpopulation involved in autoimmune pathogenesis, and their secreted cytokines, IL-2 and interferon-γ, contribute to the proinflammatory phenotype. Th17 cells enhance autoimmune response progression and can drive the autoimmune response in the absence of a concomitant Th1 response [1, 18, 29].
GC treatment has several effects on T cell subsets, including Treg expansion and Th polarization. Additionally, when treating infants, the effects of GC on thymocyte maturation must also be considered.
Effects on Regulatory T Cells
In experimental models, GCs increase the frequency of Tregs, suggesting that GC-mediated immune suppression is partially achieved through increased Treg cell number or activity [24]. Clinical studies have reported a GC-dependent increase in Treg-related cytokine expression or Treg cell number and function. In patients with asthma, GC treatment induces IL-10 synthesis, a major anti-inflammatory cytokine produced by Tregs, as well as expression of the Forkhead box P3 transcription factor (FoxP3), which is typically expressed by these cells [47]. In patients with Graves’ disease, Treg cell functionality improved after dexamethasone treatment; however, their proportion remained unchanged [44]. Treg expansion has been observed in GC-treated patients with multiple sclerosis and rheumatic disease [3, 19, 33]. Finally, GC treatment increased Treg cell number in patients with systemic lupus erythematosus (SLE) [11, 91].
There is likely more than one mechanism involved in the GC-mediated increase in Treg frequency. Unlike immature T cells, Tregs are resistant to GC-induced cell death in vivo and account for a greater proportion of T cells in mice treated with GC [23]. Moreover, expansion of tTregs by IL-10 upregulation and the facilitated generation of peripheral Tregs (pTregs) have been observed [12, 25, 75]. To this end, GCs synergize with transforming growth factor-β signaling in FoxP3 induction through induction of GILZ and consequent SMAD2 phosphorylation, which is necessary for the GC-induced increase in Treg number [15, 47, 75].
Effects on Effector T Cells
GCs affect peripheral immune responses by inhibiting or modulating effector T cell activation at several stages of the activation cascade (section “Molecular mechanisms of glucocorticoids”). Specifically, reports have described GC-mediated transcriptional regulation of TCR complex proteins [59, 64, 82] as well as modulation of TCR function and signal transduction [9, 26, 53, 65]. In particular, GCs modulate kinase expression and activity [73, 99]. Because the peripheral responsiveness of T cells appears to be regulated by the quantity and quality of intracellular signals triggered by TCR activation, the interference of GCs on TCR signaling contributes to their effect on T cells.
Another interesting GC-mediated effect is a shift in the balance of the immune response from a Th1 to a Th2 type [78]. In the initial stages of an immune response, naive cells that are committed to activation and differentiation into either Th1 or Th2 phenotypes are initially suppressed in the presence of GCs, which is consistent with their general immunosuppressive action. Indeed, GCs inhibit both T-bet, which is selectively expressed in Th1 cells, by transrepression, and GATA3, which is selectively expressed in Th2 cells, by inhibiting p38 MAPK phosphorylation. However, the greater sensitivity of T-bet to GC inhibition favors Th2 development, particularly during long-term GC treatment [49]. The GC-dependent upregulation of Itk, a Tec kinase favoring Th2 polarization, may be another polarization mechanism of GC [73].
GCs affect both Th17 polarization and function by modulating cytokines such as IL-23, IL-6, and IL-17 [39, 62]. It was also recently demonstrated that the aberrant Th17/Th1 balance in patients with SLE is linked to GC use [74]. Investigations using specific conditional knockout models will likely identify a clear correlation between Th17 and GC action.
A third GC-mediated effect on mature T cells is apoptotic cell death. The degree of activation and the timing of GC exposure (before, during, or after activation) render T cells sensitive or resistant to GC-induced apoptosis [105].
Effects on Thymocytes
GC actions in thymocytes have been the subject of several studies. However, several controversies remain. It has long been known that high systemic GC levels lead to thymus involution whereas low concentrations result in oversized thymus. During development, immature double-negative thymocytes (CD4−CD8−) progress to the double-positive phenotype (CD4+CD8+) and undergo positive or negative selection depending on mild or strong binding, respectively, between their major histocompatibility complexes and self-antigens. At this stage, double-positive thymocytes are highly sensitive to GC-induced apoptosis that, in turn, can be antagonized by TCR signaling and vice versa (termed mutual antagonism) [4]. A number of genetically modified mouse models with altered GRs have been produced to help understand the role of GCs in thymocyte death by neglect. These studies have yielded conflicting results. It seems reasonable to conclude that GR triggering is dispensable for thymocyte development under physiological conditions but may affect thymocyte turnover under stress conditions, such as infection, or during prolonged GC treatment [77, 102]. This topic has been previously discussed in depth [81].
Effects on the Cells of Innate Immunity
Adaptive immunity is not the only player in autoimmunity. In fact, innate immune system cells, including dendritic cells (DCs), natural killer (NK) cells, NK T lymphocytes, and macrophages, also modulate self-tolerance. Epithelial and endothelial cells have also been implicated in the pathogenesis of autoimmune diseases. GCs are able to influence the function of these cells and were recently found to affect TLR signaling [68], thus contributing to the counteraction of autoimmune diseases.
GC-treated immature DCs are unable to undergo full maturation and prime Th1 cells efficiently [56]. After GC exposure, mature DCs show a decreased ability to present antigens and activate T cells and are reprogrammed into the so-called tolerogenic DC. At the molecular level, such an orientation to tolerance is driven, at least in part, by GILZ upregulation, which can prevent the expression of CD80, CD86, and CD83 as well as DC production of inflammatory chemokines. This interferes with the NF-κB pathway, which allows for DC maturation, and simultaneously induces IL-10 production [41, 80]. GC-treated DCs also possess the capacity to convert CD4 T cells into IL-10-secreting Tregs, potently suppressing the proliferation of responder T cells [97].
During inflammatory processes, macrophages resolve inflammation in several ways, including recognition and phagocytosis of apoptotic cells (such as dead neutrophils). Endogenous GCs can exert distinct effects on macrophages depending on their concentration and the functional status of the macrophages. Some studies suggest that GCs reduce rolling, adhesion, and transmigration of proinflammatory monocytes by lowering the expression of adhesion molecules such as β2 integrins [lymphocyte function-associated antigen-1 (LFA-1) and macrophage-1 antigen (Mac-1)] and selectins, which interact with their endothelial counterparts on endothelial cells [51]. Other studies suggest that GC treatment promotes migration of anti-inflammatory monocytes, inducing their differentiation toward specific subtypes with particular anti-inflammatory phenotypes [37]. Moreover, GCs exert immunostimulatory effects on macrophages favoring cell uptake [51, 106]. Interestingly, this occurs even in alveolar macrophages, in which cell uptake is inhibited by the lung environment [57].
Acute cortisol that is released following stress or intravenous GC injection causes a profound depletion of eosinophils and basophils due to apoptosis, thus preventing tissue invasion. By contrast, systemic GCs elicit opposing effects on neutrophils by increasing the number of peripheral neutrophils, as well as their bone marrow progenitors [96], and delaying spontaneous apoptosis. Notably, GCs reduce reactive oxygen species generation and proinflammatory cytokine expression in peripheral neutrophils [43].
Stress decreases NK cell activity, and recently the molecular mechanisms of GR-mediated gene suppression have begun to be unraveled in these cells [20]. However, a recent report describes a synergistic effect of GCs with IL-15 in increasing the proliferation of human peripheral blood NK cells and protecting against cytokine-induced cell death. This is an emerging topic because of the potential use of NK cells as a powerful tool in cancer immunotherapy [63].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


