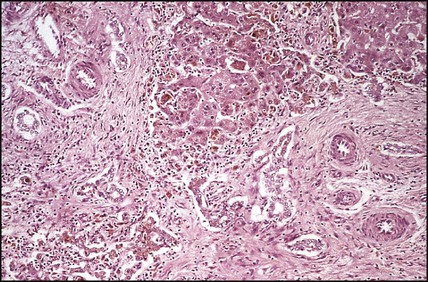
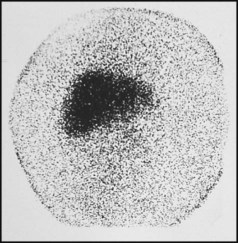
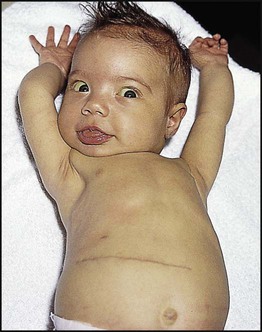
Features of liver disorders in children are: • Prolonged (persistent) neonatal jaundice is the most common presentation of liver disease in the neonatal period. • The earlier in life biliary atresia is diagnosed and treated surgically, the better the prognosis. • Transmission of hepatitis B surface antigen-positive mothers is prevented by immunising their babies. • Chronic liver disease (Fig. 20.1), cirrhosis and portal hypertension are uncommon and should be managed by, or in conjunction with tertiary or national centres. • Liver transplantation is an effective therapy for acute or chronic liver failure, with a >80% 5-year survival rate; it is only performed at national centres. Many newborn infants become clinically jaundiced. About 5–10% are still jaundiced at >2 weeks of age (3 weeks if preterm), when it is called ‘prolonged (or persistent) neonatal jaundice’. This is usually an unconjugated hyperbilirubinaemia, which resolves spontaneously (Box 20.1). Prolonged neonatal jaundice caused by liver disease is characterised by a raised conjugated bilirubin (>20 µmol/L) and is usually accompanied by: In neonatal hepatitis syndrome, there is prolonged neonatal jaundice and hepatic inflammation. Its causes are listed in Box 20.1, but often no specific cause is identified. In contrast to biliary atresia, these infants may have intrauterine growth restriction and hepatosplenomegaly at birth. Liver biopsy (Fig. 20.6) is often non-specific, demonstrating a giant cell hepatitis. Case History • Prothrombin time – 28 s (normal range 10–13 s) • Serum bilirubin 178 micromol/L – 140 micromol/L conjugated. The investigation of conjugated hyperbilirubinaemia is shown in Figure 20.2. The TIBIDA radionuclide scan showed no excretion at 24 h (Fig. 20.3) and a liver biopsy suggested biliary atresia (Fig. 20.4). A hepatoportoenterostomy was performed at 6 weeks of age (Fig. 20.5).
Liver disorders
Neonatal liver disease
Neonatal hepatitis syndrome
α1-Antitrypsin deficiency
Liver disorders
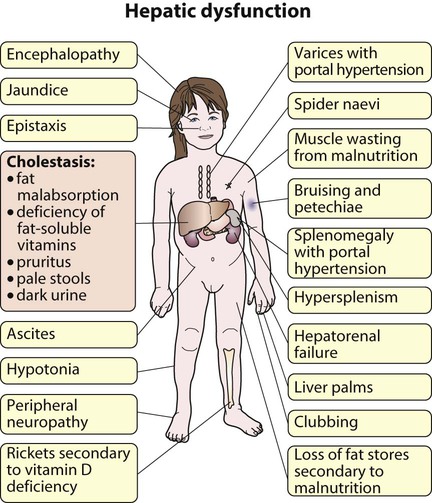
 In prolonged (persistent) neonatal jaundice, check if it is conjugated hyperbilirubinaemia, as this is due to liver disease.
In prolonged (persistent) neonatal jaundice, check if it is conjugated hyperbilirubinaemia, as this is due to liver disease.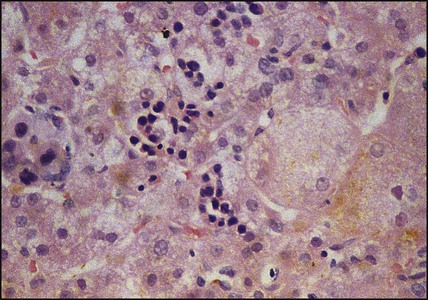
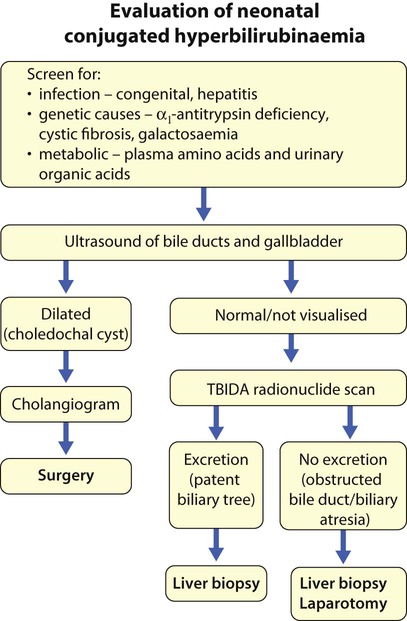
 In persistent jaundice, always look to see if the stools are pale – suggests bile duct obstruction.
In persistent jaundice, always look to see if the stools are pale – suggests bile duct obstruction.


