Fig. 11.1
MRCP of a 17-year-old patient with PSC. Note the intrahepatic and extrahepatic beading of the biliary tree with sequential strictures and saccular dilatations. The common bile duct wall is also thickened
Table 11.1
The Majoie classification of cholangiographic findings in primary sclerosing cholangitis [15]
Intrahepatic | Extrahepatic |
|---|---|
Minimal strictures with normal diameter of biliary ducts or minimal dilatation | Duct contours have slight irregularities without stenosis |
Multiple strictures with saccular dilatations and reduction of intraparenchymal arborization | Segmental stenosis of the biliary duct |
Lack of visualization of one of the main hepatic ducts | Almost the entire biliary duct is stenotic |
Pseudo-diverticular out-pouching of the biliary ducts with irregular duct margins or diameter |
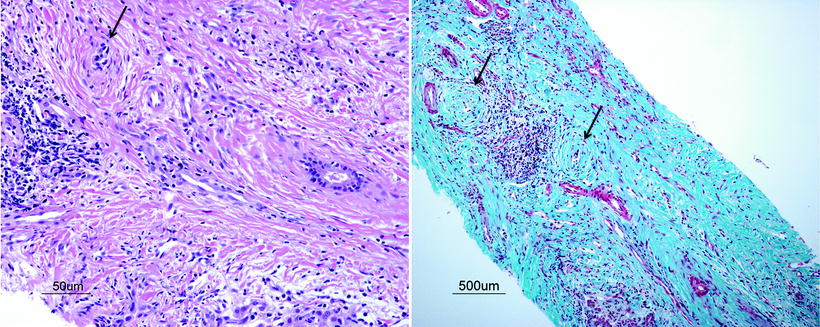
A liver biopsy is indicated in a child with IBD and persistently raised liver enzymes, particularly if the GGT is also elevated. The liver biopsy should be performed in this setting regardless of the radiological findings as PSC may involve the smallest biliary ducts and only be detected microscopically. A biopsy is also needed to identify any evidence of ASC, which requires a different therapeutic approach (see section “Autoimmune Sclerosing Cholangitis”). The histopathology in small duct PSC can reveal acute and/or chronic cholangitis, fibrosis, or cirrhosis (Table 11.2) [19]. A finding known as “onion skinning” is the most characteristic histopathologic lesion of PSC, which describes concentric periductal fibrosis (Fig. 11.2).
Portal tract fibrosis with cholangitis and portal hepatitis |
Periportal fibrosis or hepatitis |
Septal fibrosis and/or bridging fibrosis |
Cirrhosis |
Fig. 11.2
Liver histopathology from a 15-year-old boy with PSC and UC. On H&E staining (a) and trichrome staining (b) there is evidence of periductal fibrosis (“onion skinning,” arrow) with degeneration and atrophy of the ductal epithelium of some cholangioles. Also note the severe bridging fibrosis with fibrous septae linking portal tracts. (Images courtesy of Dr. Ernest Cutz, MD, FRCPC, Hospital for Sick Children, University of Toronto)
Treatment of Primary Sclerosing Cholangitis
Unfortunately, pharmacological treatments for PSC have been disappointing, as they have not shown a decrease in the rate of disease progression or a change in the rate of survival. Treatment with corticosteroids, penicillamine, and azathioprine have not shown long-term success in improving outcomes in PSC [20, 21]. Tacrolimus has been shown to cause a reduction in ALT and ALP but was poorly tolerated in adults [22]. Mycophenolate mofetil, methotrexate, budesonide, etanercept, pentoxifylline (an anti-TNF agent), and pirfenidone (an antifibrotic agent) have all not shown any short- or long-term benefit [22]. Ursodeoxycholic acid (UCDA), a dihydroxy bile acid, has been the mainstay of medical therapy for PSC, having been shown to decrease the hepatotoxicity of other bile salts, provide a hepato-protective effect, and improve the serum ALT [23, 24]. However, a recent trial of high dose ursodeoxycholic acid (28–30 mg/kg/day) vs. placebo in adults revealed an association with higher rates of liver transplantation and death, suggesting toxicity at these doses [22]. The AASLD guidelines recommend against the use of UCDA in adults with PSC; however, there is insufficient evidence to establish firm guidelines for or against the use of this therapy in children [1]. Many pediatric hepatologists continue to utilize ursodeoxycholic acid at lower doses (up to 20 mg/kg/day divided twice daily), as there may be potential benefit for improving outcomes. Oral vancomycin, at 50 mg/kg/day, has also been reported to improve liver biochemistry and reduce inflammation on liver biopsy in children with both UC and PSC [25]. Of note, those with liver cirrhosis on biopsy did not show a statistically significant degree of improvement. Furthermore, survival outcomes after long-term vancomycin use have not been reported. Minocycline and metronidazole have also been shown to reduce abnormal liver biochemistry in PSC [22]. These data suggest that antibiotics can decrease inflammation in the PSC liver, implicating bacteria in the pathogenesis of PSC. Overall, the management of PSC involves close co-operation with a hepatologist, limitation of exposure to hepatotoxic medications, as well as regular monitoring of liver biochemistry.
While MRCP is a noninvasive diagnostic tool, which is desirable in pediatrics, an ERCP has the advantage of providing therapeutic potential. In the setting of a localized large duct stricture, a biliary intervention via ERCP can ameliorate the course of disease. A bile duct stricture in PSC can lead to worsening liver function with progression to cirrhosis as well as increased risk of bacterial cholangitis [26]. With ERCP-mediated balloon dilatation ± stenting of a stenotic duct, there can be improvement of liver biochemistry, including bilirubin, decreased jaundice and pruritus (if present), and improved survival [27].
Outcomes in Primary Sclerosing Cholangitis
Primary sclerosing cholangitis causes liver fibrosis and can progress to liver cirrhosis, as well as hepatic failure. Angulo et al. in 2002 compared patients with small-duct PSC (N = 18) vs. classic PSC (N = 36), and found that survival with native liver was significantly higher in the group with small duct PSC (p = 0.04), suggesting a better prognosis with this pattern of disease [28]. A small group of patients (3/18) with small-duct disease were found to develop classic PSC during follow-up. It is unclear if the two patterns of disease are on a continuum or are distinct entities. In a single-centre study from Mount Sinai, 19% of children required liver transplantation [9]. A recent study from the SPLIT (Studies of Pediatric Liver Transplantation) database assessed the outcomes of children with PSC following liver transplantation [29]. The patient survival was 98.7% at 1 year and 86.6% at 5 years, compared to 94.3 and 88.2%, respectively, in the non-PSC group. Meanwhile, the graft survival was 93% at 1 year and 76.1% at 5 years, compared to 90 and 79.5%, respectively, in the non-PSC group. There were no significant differences in patient and graft survival between the two groups. However, with a univariate analysis, mortality was significantly higher in PSC patients with a diagnosis of IBD pretransplant. Of potential interest, a recurrence of PSC was diagnosed in 6 of 61 patients (9.8%) over a median follow-up time of 36.6 ± 32.7 months, and all who recurred had been diagnosed with concurrent IBD pretransplant. In a prospective comparison of adult PSC patients with a dominant stricture of the biliary duct, the transplant-free survival after 18 years of follow-up was poorer in the group of patients with concomitant UC (23 vs. 77.8%, respectively, p = 0.045) [30]. Proctocolectomy has not shown any difference in survival or decrease in the complications of portal hypertension, in adult patients with concomitant PSC/UC [31]. There is insufficient evidence to recommend colectomy in children with PSC/UC to prevent progression of PSC disease or liver transplantation.
Another important complication of PSC is an increased risk of cholangiocarcinoma. In a study of 1,274 adult patients with UC, the overall prevalence of this malignancy was 0.3%; however, in patients with concurrent PSC, the prevalence was 13% [32]. Furthermore, patients with PSC and a dominant bile duct stricture have a statistically significant increased risk of cholangiocarcinoma, as well as colorectal carcinoma, if they have concurrent IBD [30]. These cancers are rare in children, and the AASLD guidelines do not recommend increased surveillance in the pediatric population with PSC/IBD, while adult screening with colonoscopy is recommended every 1–2 years after diagnosis [1].
Other Autoimmune Liver Diseases
Autoimmune Hepatitis
AIH is an autoimmune disorder characterized by hepatic inflammation and it can be categorized according to the type of autoantibodies present. In AIH type 1, antinuclear antibodies and/or smooth muscle antibodies (ANA/SMA) are frequently positive, while in AIH type 2 antibodies to liver/kidney microsomal type 1 (LKM1) can be positive. Children can be seronegative, or have antibodies titers that are lower than in adults such that 1:20 for ANA or SMA, and 1:10 for anti-LKM1 are significant levels in children, while in adults 1:40 is clinically relevant, as per AASLD guidelines [33]. Both types of AIH favor a female predominance (75%), have elevated immunoglobulin G (IgG) titers, and in children, progress with a similar disease course. However, patients with type 2 disease frequently present at a younger age (median age: 7.4 vs. 10.5 years) [34]. The clinical presentation can include nonspecific symptoms such as fatigue, nausea, abdominal pain, and arthralgia, while others may present with jaundice, acute hepatitis, or even liver failure [33]. A liver biopsy is necessary to confirm the diagnosis as well as to rule out other causes of liver biochemistry derangement. Findings include portal tract inflammation, lobular hepatitis, interface hepatitis, and cirrhosis [34]. Plasma cell infiltration of the portal tracts are commonly associated with the periportal hepatitis; however, in some patients with AIH this may be absent [33]. Autoantibodies are integral to making a diagnosis of AIH as described earlier. However, ANCA can also be frequently positive in AIH type 1. In one study within adults, 65% of 46 patients with AIH type 1 were ANCA positive; however, none of the 19 patients with AIH type 2 were positive [35]. The International Autoimmune Hepatitis Group (IAIHG) developed and revised a scoring system for the diagnosis of AIH in 1999 that incorporates many of these parameters [36]. Some indices included in the score are liver biochemistry, autoantibodies, viral serology status, drug or alcohol use, and findings on liver histology. With a total AIH score above 15 points, the diagnosis of AIH is considered definite, while with a score between 10 and 15 points, AIH is probable. Ebbeson and Schreiber retrospectively evaluated this tool in 2004, in children with diagnoses of AIH (N = 21) and sclerosing cholangitis (N = 7) [37]. They found 85.7% (18/21) of patients clinically diagnosed with AIH to have “definite AIH” according to the IAIHG tool, while 14.3% (3/21) had “probable AIH.” Of the patients with sclerosing cholangitis, all four with isolated cholangitis were found to have a score of <10 points, while the three patients with overlap disease (ASC) had scores consistent with definite AIH. This study evaluated a small group of patients; however, it suggests that patients with hepatic inflammation consistent with AIH may be reliably differentiated from isolated PSC with this tool. However, this tool is not specific enough to rule out ASC in patients who appear to have AIH.
Immunosuppressive therapy is the standard of care for AIH in children due to the increased disease severity at onset as well as the aggressive course with poorer long-term outcomes observed if treatment is delayed [33]. Prednisone (1–2 mg/kg/day) is used to induce remission and can be continued at a lower dose (2.5–5 mg/day) for long-term maintenance. Azathioprine is usually added for maintenance treatment, as recommended in AASLD guidelines [33]. In children who fail to respond to these medications, additional options include MMF, cyclosporine, and tacrolimus, although the evidence for use of these agents is based on case series and not randomized trials comparing to standard treatment regimens [33]. After successful therapy for 2–3 years, with persistent normalization of liver biochemistry, immunosuppression discontinuation may be attempted after a liver biopsy has confirmed complete histologic remission of disease.
Other autoimmune diseases, such as hypothyroidism, occur in 20–22% of patients with AIH [34]. Of particular relevance here is the overlap with IBD. Within the IBD population, AIH is an uncommon finding: 0.9% of 1,009 children in a multicenter study with IBD, and 1.4% with UC had a “chronic active hepatitis.” [7] However, within the pediatric AIH population, IBD is more frequently diagnosed with a prevalence of 12–20% of children with AIH in one study, advocating a low index of suspicion for IBD in this population [34] (Fig. 11.3).
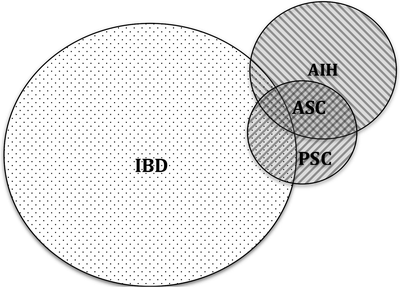
Fig. 11.3
Overlap between IBD and chronic liver diseases. This Venn diagram represents the relationship between IBD and chronic liver diseases, as well as the perceived prevalence (not drawn to scale)
Autoimmune Sclerosing Cholangitis
Autoimmune sclerosing cholangitis occurs almost exclusively in children and young adults [19, 38, 39]. It is diagnosed when features of both PSC and AIH are present on liver biopsy and imaging; however, as in PSC, there are no specific diagnostic criteria. These patients have an increased frequency of concomitant IBD (44%) compared with both PSC and AIH [34]. In one study at King’s College in London, children referred with biochemical evidence of liver disease as well as positive autoantibodies were prospectively assessed [19]. Of the 76 patients referred between 1984 and 1997, 55 patients, who were investigated with both a liver biopsy as well as cholangiography, were included in the study. A diagnosis of ASC was attributed to patients who had histological evidence of AIH and cholangiographic evidence of intrahepatic or extrahepatic sclerosing cholangitis. Of note, this definition of ASC may have precluded a diagnosis of small duct sclerosing cholangitis, overlapping with AIH, which would only be diagnosed histologically. Nonetheless, 27/55 (49%) patients were diagnosed with ASC while 28/55 (51%) of patients were diagnosed with AIH. There were no significant differences in the median age at diagnosis (10.5 years ASC, 11.8 years AIH) or female preponderance (55% ASC, 79% AIH). Signs and symptoms were also similar at diagnosis with the majority presenting with jaundice, hepatomegaly, and splenomegaly, except for pruritus, which was significantly increased in AIH (25 vs. 7% in ASC). Laboratory investigations at baseline were mostly similar between ASC and AIH except for median AST (102 IU/L in ASC, 333 IU/L in AIH, p = 0.002), median total bilirubin (1.2 mg/dL or 20 μmol/L in ASC, 2.0 mg/dL or 35 μmol/L in AIH, p = 0.04), and ANCA positivity (74% in ASC, 36% in AIH, p = 0.009). In children with ASC, ANA and SMA titers were positive in 20/27 (74%) of patients and LKM1 antibodies were present in 1/27 (4%) of patients. Autoimmune disorders in first-degree relatives were also significantly increased in AIH (71 vs. 37% in ASC). On histology, there was increased acute and/or chronic cholangitis in patients with ASC compared to AIH (35 vs. 12%, respectively, p = 0.049) but decreased portal tract inflammation (58 vs. 92%, respectively, p = 0.004).
Routine sigmoidoscopy with rectal biopsies was also performed in all patients within this cohort except for one who refused. IBD was diagnosed in 44% of patients with ASC. This was significantly different from the 18% of patients with AIH who were also diagnosed with IBD. There was potential for the underestimation of the prevalence of IBD in this population as the colitis associated with PSC frequently has rectal sparing and only rectal biopsies were taken in this cohort.
On treatment with immunosuppression, 100% of patients with AIH experienced normalization of liver biochemistry, while in ASC, 83% had normal AST levels, and 73% had normal bilirubin. Meanwhile, there was no significant difference in rate of relapse with 36% of AIH patients versus 33% of ASC patients experiencing at least one episode of relapse. Of those patients with ASC who had follow-up cholangiography, 8/17 had progression of intrahepatic and extrahepatic biliary disease. Upon follow-up, all patients were alive and no patients with AIH required liver transplantation in this cohort. Meanwhile, 4/27 (15%) of children with ASC required liver transplantation. This study suggests that children with AIH/IBD should be screened with cholangiographic imaging to assess for PSC, as this could have implications for a poorer prognosis [19]. This is clearly stated in the AASLD practice guideline in 2010 [33]. Conversely, children with cholangiographic evidence of PSC should undergo liver biopsy and autoantibody detection to screen for evidence of overlap, as ASC requires immunosuppressive treatment [1, 33]. According to AASLD guidelines, though based on limited evidence, treatment with corticosteroids (±azathioprine) may be attempted in PSC patients with elevated IgG, positive autoimmune antibodies, and interface hepatitis on liver biopsy.
Drug Hepatotoxicity
Methotrexate
Methotrexate, an anti-inflammatory that inhibits synthesis of purines and pyrimidines via inhibition of the folate metabolism, has been found to maintain remission in IBD [40]. Previous use in rheumatological diseases has identified a hepatotoxic effect of methotrexate, with reports of elevation of liver biochemistry and hepatic fibrosis (15% prevalence) [41]. The use of methotrexate in psoriasis came with the recommendation in 1996 to perform protocol liver biopsies after administration of a cumulative dose of 1,500 mg [42]. With further investigation, this practice was changed. Updated publications for psoriasis and juvenile idiopathic arthritis have included recommendations for biopsies in cases of refractory abnormal liver biochemistry despite decreased or held doses of methotrexate. A biopsy can also be considered with higher cumulative doses of methotrexate (3.5–4.0 g) [43, 44]. Subsequent studies in adults and children with IBD have also not supported protocol liver biopsies although the numbers of patients in each study were limited. The two adult retrospective studies that included the most number of liver biopsies after methotrexate administration (Fournier 2010: N = 17/87 and Te 2000: N = 20/32) demonstrated that an increased cumulative dose of methotrexate did not correlate with worse liver histopathology [45, 46]. In the pediatric IBD literature, retrospective studies of methotrexate use have described abnormal liver biochemistry requiring dose reductions or drug discontinuation (Turner 2007: N = 7/60 and Uhlen 2006: N = 2/61) [47, 48]. However, liver biopsies were not performed in these cases making it difficult to interpret the exact effect of methotrexate in children with IBD. Previous studies of methotrexate hepatotoxicity have also described hepatic steatosis on liver biopsy. These findings were significantly worse in psoriatic adult patients with obesity ± diabetes mellitus compared to psoriatic patients without these risk factors (and matched for cumulative methotrexate doses) [41, 49]. It is rare for patients to receive baseline liver biopsies prior to methotrexate use. Therefore, the contribution of methotrexate to the degree of steatosis is unclear. Nonetheless, patients with obesity or diabetes mellitus require close monitoring as methotrexate could further exacerbate underlying nonalcoholic fatty liver disease (NAFLD). From the available limited evidence, when starting methotrexate, liver biochemistry should be obtained at baseline and in follow-up: weekly after the initial dose or after changes in doses for the first month, and every 2–3 months thereafter. With development of persistently abnormal liver biochemistry, in consultation with a hepatologist, the dose of methotrexate can be adjusted, or temporarily held with mild to moderate abnormalities (e.g., up to 2–3× ULN, the upper limit of normal), or completely discontinued if highly abnormal (e.g., >5× ULN). Liver biopsies can be reserved for cases of persistently abnormal liver biochemistry or if discontinuation of the methotrexate would be deleterious for the IBD management. Caution should be exerted in prescribing methotrexate to patients with underlying liver disease and avoided in children with PSC unless under exceptional circumstances (Table 11.3).
Table 11.3
Drug hepatotoxicity reported in different IBD therapies
Drug | Liver outcome |
|---|---|
Methotrexate | Hepatic fibrosis |
Hepatic steatosis | |
Azathioprine/6-Mercaptopurine | Hepatitis |
Peliosis hepatis | |
Veno-occlusive disease | |
Severe cholestasis | |
Nodular regenerative hyperplasia | |
Portal hypertension | |
5-aminosalicylic acid | Fever, rash, jaundice, hepatomegaly |
Acute liver failure | |
Anti-TNF, biologic agents | Hepatitis |
Hepatitis B reactivation | |
Hepatosplenic T-cell lymphoma | |
Cyclosporine | Mild elevated liver biochemistry |
Biliary sludge |
Azathioprine/6-Mercaptopurine
The thiopurine immunomodulators provide an anti-inflammatory effect, and maintenance of remission in IBD, by conversion to 6-thioguanine nucleotides, which insert into the DNA of rapidly dividing cells and suppress replication [50]. While, adverse effects can occur in 15–30% of patients with IBD on thiopurines, hepatotoxicity is rare [51]. Adult retrospective studies have shown frequencies of Azathioprine (AZA)/6-Mercaptopurine (6MP) hepatotoxicity ranging from 0 to 5.2% [52–57]. The majority of patients improved with dose reductions or drug discontinuation. One patient developed peliosis hepatis, blood-filled cavities on histopathology, which resolved with drug withdrawal. Other reports have identified isolated cases of veno-occlusive disease and severe cholestasis, of which both resolved upon drug discontinuation [58, 59]. A case–control study of adults with IBD treated with azathioprine identified 37 cases of nodular regenerative hyperplasia and found a positive association with male gender and stricturing IBD disease. These cases were identified either due to abnormal liver biochemistry versus symptoms or abdominal imaging consistent with portal hypertension (PHT). Of these cases, 15 required treatment for PHT and 1 patient received a liver transplant [60]. Meanwhile, studies in children have assessed smaller cohorts of patients: Cuffari et al. in 1996 found 1 in 25 adolescents (4%) developed hepatitis, confirmed with liver biopsy [61]. Grossman et al. in 2008 assessed a cohort of 30 children <6 years of age who were prescribed elevated doses of azathioprine (3 mg/kg/day) due to suspected differences in pharmacokinetics in this age group. Two patients (6.7%) developed abnormal liver biochemistry that resolved without dose discontinuation. Both these patients had normal levels of thiopurine methyltransferase (TPMT), the enzyme implicated in toxicity when deficient [62]. Another study of 22 children with IBD by Kader et al. in 2000 also found that TPMT levels did not correlate with hepatotoxicity in the 2 children who developed hepatitis [63]. A dose monitoring strategy for thiopurine administration may include liver biochemistry at baseline, and then weekly for the first 4 weeks of therapy, twice monthly for the following second and third months, and then monthly [64].
Other IBD Therapies and the Liver
Other IBD treatments have reported rare cases of hepatic disturbance. Within the 5-aminosalicylic acid class of medications, sulfasalazine has been associated with significant cases of fever, rash, jaundice, hepatomegaly that either progressed to acute liver failure, or resolved with corticosteroid administration [65, 66]. These cases were suspected to be idiosyncratic reactions to the sulfa moiety within this compound.
Hepatic steatosis can occur with glucocorticoid administration due to increased lipogenesis and decreased free fatty acid oxidation pathways [67]. Children with IBD who are treated with corticosteroids are at risk of this complication. However, despite frequent use of this medication, there are few, if any, biopsy-proven case reports of this complication in adults or children [68]. This likely indicates that the short courses of steroids used in IBD do not frequently cause significant liver disease.
Hepatotoxicity with anti-TNF use is rare. However, after 35 cases of hepatitis, cholestasis, and acute liver failure were reported to the FDA MedWatch program, in patients using anti-TNF agents, a black-box warning was issued to alert physicians to the possible associated hepatotoxicity [69]. Also concerning were the findings of increased hepatitis B reactivation in patients on these biologic agents. It is possible that endogenous TNF promotes viral clearance and its loss contributes to the increased viral replication [69]. Cyclosporine was studied in 111 patients with IBD in 2008 and 21 (19%) were found to develop mild elevations in liver biochemistry that did not cause any adverse effects. Only one patient was investigated further and was found to have biliary sludge on ERCP [70].
Finally, total enteral nutrition (TEN) is a therapy that has been used to both induce and maintain remission in Crohn disease. This treatment is preferred to the use of total parenteral nutrition (TPN) due to a more physiologic delivery of nutrition as well as a lower side effect profile thus avoiding the feared intestinal failure-associated liver disease. Two previous reports have studied the liver biochemistry profile in TEN for IBD: Dolz et al. in 1989 did not find any statistical differences between IBD patients on TEN vs. not on TEN (though this was not randomized). Meanwhile, Schtorje and Hoekstra in 2010 found a transient increase in liver biochemistry in the first 6 weeks of treatment, that subsequently normalized spontaneously without progression to liver disease, over a mean follow-up period of 2.1 years [71, 72].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


