Invasion, Metastasis, and Angiogenesis
SHARON ROMANO-FITZGERALD  ALEXANDRE BUCKLEY DE MERITENS
ALEXANDRE BUCKLEY DE MERITENS  ANGELES ALVAREZ SECORD
ANGELES ALVAREZ SECORD  ELISE C. KOHN
ELISE C. KOHN
INTRODUCTION
Gynecologic cancers are potentially curable if diagnosed at an early stage, when the tumor remains confined to the primary organ; survival is compromised by the dissemination or metastasis of the disease to vital organs at more advanced stages. In ovarian cancer, the leading cause of death for gynecologic malignancies in the United States, survival rates decrease dramatically with advanced disease presentation. The 5-year relative survival rate in advanced ovarian cancer with spread outside the pelvis is 26.9%, in contrast to a rate of 91.5% in stage I disease. A similar pattern is seen in cancers of the cervix and uterine corpus, with 5-year relative survival rates for localized disease of 90.7% and 95.4%, respectively; for distant disease, the rates are 16.2% and 16.4%, respectively (Howlader, et al: http://seer.cancer.gov/csr/1975_2009_pops09). The progression from hyperplasia to carcinoma in situ to invasive and then metastatic cancer determines staging and prognosis (Fig. 4.1).
Metastasis, the spread of malignant cells from the primary tumor to other sites, requires tumor cells detach from their primary mass, invade the stroma, travel to a secondary site via the lymphatic and/or the vascular system, extravasate and migrate within the secondary stroma, and grow (1). Tumor cells must also interact closely with elements in their microenvironment and communicate with them to successfully achieve metastasis (2, 3). The peritoneum, omentum, and mesenteric serosa are permissive environments to which the tumor cells attach and grow, co-opting support cells and vasculature to sustain their survival and growth. Through overcoming anoikis, a form of apoptosis triggered by loss of attachment to the solid extracellular matrix (ECM) substratum, cancer cells survive in the intra-abdominal milieu and in the peritoneal fluid or ascites (4, 5).
The endothelium is the cellular lining on the luminal side of blood vessels, which serve as the first docking interface for circulating tumor cells. The endothelium creates a semi-permeable barrier sitting over its basement membrane; together they control blood–tissue exchanges of fluids, nutrients, and metabolic wastes, while preventing access to and from the tissues for pathogens and tumor cells (6). Altering endothelial cell–cell adhesion alters this critical endothelial barrier function. Loss of cell–cell adhesion allows endothelial cells to constrict in response to noxious stimuli. Constriction permits retraction, leaving acellular regions of sub-endothelial basement membrane exposed. Tumor cells attach to and degrade these denuded matrix regions (7).
The primary tumor and metastatic sites also have a high metabolic demand requiring adequate blood supply, absent which, tumor cells survive by autophagy, internal protein degradation, and recycling (8, 9). Angiogenesis, the formation of new vessels, is necessary both as a route for metastasis and to provide the metastatic sites with exogenous nutrients to support tumor growth (1). Many proangiogenic and antiangiogenic factors are secreted into and by elements of the tumor microenvironment leading to new vasculature that supports tumor survival and progression (10). Invasion, angiogenesis, and metastasis are thus orchestrated within the tumor microenvironment through a dynamic interaction between the tumor cells, the ECM, stromal and immunologic cells, and secreted chemokines and growth factors (1,2). Cancer cells use normal cellular processes to survive and progress, albeit without the tight regulation seen in normal cells. Tumor cells adapt in response to environmental and cellular stress and upregulate different mechanisms as cell migration, angiogenesis, autophagy, and apoptosis for their survival (11).
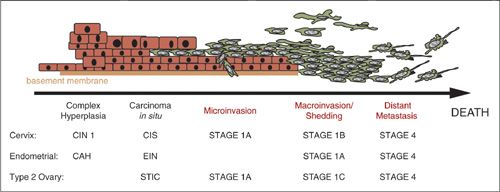
FIGURE 4.1. Paradigm of cancer progression. Progression from the earliest detectable event of complex hyperplasia to overt invasive and metastatic malignancy is over a decade for endometrial and cervical cancers. The new recognition of the tubal epithelium as the source for type 2 high-grade serous ovarian cancer leaves this time frame still undetermined. Where identified early, each of these cancers has a high cure rate and long overall survival. However, when identified at or beyond the stage of microinvasion, progressive disease and death from disease is common.
PATTERNS OF SPREAD
Gynecological cancers are able to disseminate in a multitude of ways (Fig. 4.2), and morbidity and mortality are associated with extent of tumor spread, invasion, and metastasis. The classical mechanism of dissemination of solid tumors is invasion of surrounding tissues followed by subsequent hematogenous and/or lymphatic spread. Endometrial and cervical cancers tend to be found as solid disease that extends locally and/or spreads in a defined pattern through the lymphatics, and later through the vasculature. Their staging is dependent on the extent of local structural invasion and/or lymph node status. In most solid tumors, such as these gynecologic cancers and breast cancer, death is usually the consequence of distant metastasis.
In ovarian and fallopian tube carcinomas, primary and early dissemination occurs through the shedding of tumor cells (2,12) directly into the abdominal cavity; these cells then spread via the passive movement of peritoneal fluid. Cancer cells in the peritoneal fluid or ascites survive in this hypoxic, structure-free milieu by resisting anoikis (13). Attachment to serosa or peritoneum precedes local invasion and secondary metastasis; the presence of lymph node disease in ovarian cancer indicates invasion, involvement of the next step after shedding. Vascular dissemination for these and peritoneal serous cancers is a late event leading to parenchymal organ metastasis. Therefore, the ability of a tumor to spread or shed is not an isolated process; it requires upregulation of survival mechanisms as well as close interaction with other cells and the local environmental milieu (2,14).
MICROENVIRONMENT
The genetic and phenotypic make-up of a tumor is a major determinant of its metastatic efficiency, and a receptive microenvironment is a prerequisite for successful tumor growth (15,16). Sir James Paget stated in 1889 that the microenvironment of each organ, the soil, influences the survival and growth of tumor cells, the seed (16,17). Multiple overlapping signal and adhesion networks cooperate to enable molecular and structural remodeling of tissues that overall support tumor invasion, growth, and metastatic dissemination. Cellular behavior and the activation or inactivation of genes are influenced heavily by the microenvironment (1,14). Gene expression profiling and proteomics have identified a variety of factors that are upregulated in the microenvironment of gynecological tumors. This reinforces the idea that complex interplay of molecules and signals are needed for tumor sustainability. Additionally, the microenvironment has a plasticity that allows participating elements to vary throughout the events of cancer progression. The flexibility of the microenvironment supports survival of the tumor cells and their ability to withstand an otherwise harsh environment with elements of hypoxia, metabolic stress, and inflammation (3).
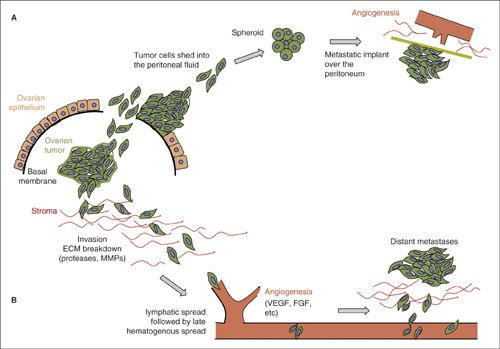
FIGURE 4.2. Ovarian cancer: shedding and spreading. The primary ovarian tumor located in the ovarian stroma metastasizes in two different ways. The first mechanism (A), occurring early is by shedding cancer cells into the peritoneal cavity after reaching the ovarian surface; the second mechanism (B), later occurring, is invasion into surrounding tissues. Cells shed in the peritoneal fluid, form spheroids that have acquired mechanisms to resist anoikis. These spheroids will then implant on the peritoneal mesothelial surface and cause stromal activation with desmoplasia, inflammation, and angiogenesis. ECM: extra-cellular matrix; FGF: fibroblast growth factor; MMPs: matrix metalloproteinases; VEGF: vascular endothelial growth factor.
Inflammation/Priming
Inflammation may promote carcinogenesis and dissemination through several mechanisms affecting growth, invasion, angiogenesis, and neoplastic changes (18). These inflammatory mechanisms include but are not limited to the production of reactive oxygen species and free radicals that directly damage DNA and proteins, and production of proinflammatory/proangiogenic molecules, such as cytokines, tumor necrosis factor-α (TNFα), and interleukins (18). Various immune cells have been found as part of the tumor inflammatory infiltrate. Among them, macrophages play a central role in most solid malignancies (19). Some T cells facilitate invasion of tumor cells by altering macrophages into a more invasive phenotype in an IL-4 dependent manner supporting the idea that inflammation mediators play a role in cancer progression (20). Inflammatory factors can also increase inflammatory enzymes such as cyclooxygenase-2, which lead to the production of other proinflammatory products (21). Studies have shown an inverse relationship between nonsteroidal anti-inflammatory drugs and reduced risk of ovarian and cervical cancers (18,22,23).
Multiple causes of local inflammation including exposure to talc, asbestos, and presence of endometriosis and recurrent pelvic inflammatory disease have been identified as putative risk factors for ovarian cancer (24,25). Cervical cancer studies have shown that, in addition to HPV, other sexually transmitted infections such as herpes simplex virus and Chlamydia are associated with increased risk of carcinoma in situ and/or invasive cervical cancer (26). It is believed that the subsequent inflammatory response and micro-ulceration that follows these non-HPV infections increase the epithelial cell repair and proliferation, permitting or promoting transformation (27). One study reported that cervical inflammation, independent of infections, was associated with squamous metaplasia of the cervix, a precursor to malignancy (28).
The menstrual cycle is a physiologic inflammatory process. The endometrium is a dynamic cyclic organ with cycles of growth, remodeling, differentiation, and angiogenesis. Estrogen drives these changes by facilitating the release of inflammatory mediators from the epithelial, stromal, and vascular cells of the endometrium (18,29). Increased exposure of the endometrium to inflammation occurs by the same mechanisms that are considered risk factors for endometrial cancer. These endometrial risk factors include unopposed estrogen use, polycystic ovarian syndrome, excessive menstruation, obesity, and diabetes. Therefore, it is believed that inflammation alone or in conjunction with estrogen plays a role in the development of endometrial cancer (18).
Tumor cells coopt normal host cells and rely on communication with other cell types including macrophages, T cells and other immune cells, endothelial cells, and stromal cells (20). Cancer cells may also cause transdifferentiation of host cells. Tumor-released inflammatory cytokines act on local cells to convert host fibroblasts into cancer-associated fibroblasts (CAFs). Activated CAFs help promote ovarian and endometrial tumor cell invasion through activation and production of growth factors, such as tumor growth factor-β (TGFβ) and chemokines (14,30,31). CAFs are part of a paracrine loop that increases cancer cell invasion (30). Furthermore, there are multiple cell interactions and stimulatory and inhibitory factors leading to feedback loops that help amplify and sustain the tumor cells in this environment (32).
Communication between tumor cells and host cells at distant tissue sites via circulating factors, immune cells, chemokines, and growth factors can prime and direct the location of future metastatic tumor growth (33). Tumor-associated macrophages, recruited by the presence of the tumor, alter the local chemokine balance resulting in directed migration, proliferation, and differentiation of the vascular cell network and inflammatory infiltrate of the tumor microenvironment. This functions to support tumor growth, both at the primary tumor and upon metastasis (20). Shibuya and colleagues demonstrated enhanced matrix metalloproteinase (MMP)-9, a protease that breaks down the ECM, was measured in the lung preceding demonstrable tumor cell metastasis, suggesting that this was induced by signals from the distant primary tumor in a paracrine fashion (34). Therefore, the role of the immune system in cancer is complex and supports the idea that a permissive microenvironment is critical for the initiation of tumor growth and establishment of metastatic lesions at secondary sites (15).
Chemokines/Growth Factors
Chemotactic cytokines, known as chemokines, are small, secreted proteins or peptides that help regulate cell motility. Their primary function is chemoattraction and activation of leukocytes that then recruit other immune and/or inflammatory cells from the blood to the affected site. These proteins are implicated in initiation and progression of cancer (35). Cancer cells and many cells in the malignant microenvironment produce and/or respond to chemokines in an autocrine or paracrine manner via chemotaxis, migration up a concentration gradient, or chemokinesis, migration within a chemoattractant milieu (32). Chemokines and their receptors act at many stages of tumor progression, ranging from cell transformation, angiogenesis promotion, to tumor and supporting cell growth. Tumor cell passage through the ECM, the vasculature, and/or lymphatics is mediated by chemokines (35). Growth factors, differentiated from chemokines by their definition initially as growth promoters for epithelial not inflammatory cells, also play a role in tumor ascites formation, motility, invasion, and migration (24).
One of many important chemokine pairs is the receptor CXCR4 and its ligand CXCL12. Together, they activate signaling pathways that enhance proliferation, migration, angiogenesis, and invasion of gynecologic cancers (36,38). Normal ovaries express very little CXCR4; whereas, it is expressed in approximately 60% of ovarian cancers. CXCL12 is expressed in more than 90% of ovarian cancers (37). CXCR4 was found to be the only receptor of 14 chemokine receptors studied to be expressed in ovarian cancer cells (38). Its expression is an independent negative prognostic factor for progression-free and overall survival in ovarian cancer (39). AMD3100, a CXCR4 inhibitor, blocked CXCL12-stimulated migration of ovarian cancer cells in vitro (40,41). Blockade of CXCR4/CXCL12 activation led to reduced tumor growth and prolonged survival in ovarian cancer xenografts (37).
Interleukins-8 (IL-8) and -6 (IL-6) are multifunctional chemokines that are secreted by multiple cell types, including monocytes, neutrophils, endothelial and mesothelial cells, and tumor cells. They are usually activated during an inflammatory response and function to recruit other immune cells to the affected site (42). Recent studies have shown that tumor progression and metastasis are associated with overexpression of these chemokines (43,44). Ovarian cyst fluid, effusions, blood, and tumor tissue have elevated IL-8 and IL-6 expression, correlated with poor overall survival (42,43). Bellone et al. demonstrated elevated serum levels of IL-6 in endometrial cancer patients and showed this was associated with a more aggressive and chemotherapy-resistant endometrial cancer (45). Preclinical data suggest that IL-8 and IL-6 increase resistance to chemotherapeutic treatments in ovarian cancer cells (46,47). Collectively, these observations provide rationale for targeting these chemokines therapeutically in gynecological cancers. Preliminary findings using an anti-IL-6 receptor antibody against ovarian cancer are encouraging (43).
Autotaxin (ATX) is a glycoprotein shown to have a critical role in invasive and angiogenic tumor and endothelial cell behavior (48,49). ENPP2, encoding ATX, has been identified as a drug resistance candidate gene (50,52). ATX is multifunctional inducing cell motility, angiogenesis, invasion, and metastasis in multiple models through its cleavage of lysophosphatidylcholine to yield lysophosphatidic acid (LPA), a major gynecologic cancer growth factor. Elevated expression of ATX or aberrant expression of LPA and its receptors has been found in most gynecologic cancers (48). High circulating plasma ATX concentrations are found in patients with recurrent ovarian cancer (51). Lack of specificity and sensitivity of circulating ATX concentration has precluded its application as a cancer biomarker. However, because of its important role in the development and progression of cancer, numerous studies are attempting to define an ATX inhibitor for clinical testing.
LPA, first described as ovarian cancer ascitic factor, is a growth-promoting lipid isolated from ovarian cancer patient ascites. It induces invasion and metastasis in a wide variety of cancers, including gynecologic cancers (53,56). LPA signals predominantly through G protein-coupled LPA receptors with crosstalk through inflammatory cytokine receptors and receptor tyrosine kinases. These events subsequently activate transcription factors, via the phosphotidylinositol 3-kinase pathway to propagate messages of invasion, angiogenesis, and metastasis (57,58). Wang found that the expression of LPA receptors was significantly increased in ovarian cancer tissues, compared to benign tumors and normal ovarian tissue (59,60). LPA concentrations are elevated in the plasma and/or ascites of ovarian, cervical, and endometrial cancer patients (61); however, the complexity of the relationship between the different LPAs, defined by their lipid substituents, has limited its biomarker development (57).
LPA signaling is central in the progression of most cancers (Fig. 4.3) (53,54,57,62,64). The LPA-Rho-associated kinase pathways have been involved in the course of various cancers, and inhibiting them decreases cancer invasiveness (58). Rho GTPase family members Rho, Rac, and CDC42 have roles in membrane trafficking, cell adhesion, and remodeling of the actin cytoskeleton in gynecologic and other cancers (32). Inhibition of Rho kinase significantly inhibited LPA-induced invasion and motility in human ovarian and cervical cancer cells in a dose-dependent manner in vitro and in xenograft models (58,65). Activation of these Rho kinase pathways lead to actin polymerization, followed by formation of membrane protrusions that are required for migration (32). Similar studies are under way for endometrial and cervical cancer (61).
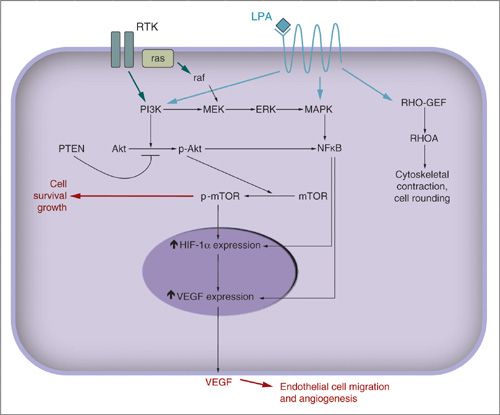
FIGURE 4.3. Molecular pathways. LPA is a well-recognized and potent prosurvival and angiogenic factor produced and secreted by ovarian cancer. LPA signals through the LPA receptor, a 7-pass transmembrane G-protein coupled receptor. LPA promotes expression of VEGF for angiogenesis and cell survival through the PI3K/AKT, NFκB, and MAPK signaling pathways. Receptor tyrosine kinases, RTK, are a broad family of receptors for multiple growth factors. Their activation also promotes cell survival, invasion, angiogenesis, and proliferation through the ras, PI3K/AKT, NFκB, and MAPK pathways. ERK: extracellular regulated kinase; MAPK: mitogen-activated protein kinase; MEK: MAP-ERK kinase; mTOR: mammalian target of rapamycin; NFκB: nuclear factor κB; Rho-GEF and RHO A are Rho family GTPases.
ADHESION
The concept of shedding and spreading in ovarian and other gynecologic cancers underscores the need for cells to attach to secondary sites and initiate secondary tumor masses. The most common sites of such adhesion is the mesothelial lining of the peritoneum, bowel serosa, or vessel walls (63,64). This occurs via adhesion molecules, of which there are four categories: integrins, cadherins, immunoglobulin super family adhesion molecules (CAMs), and selectins. Each class has different structure, functions, and binding partners, and is expressed in various degrees on the surface of different cells (Fig. 4.4). They participate in cell–cell and/or cell–substratum binding, yielding a complexity of adherence and signaling possibilities. When expression or function of adhesion molecules becomes altered by tumor progression, new signals are propagated that can promote tumor growth, survival, and metastasis (14,66).
Cell–Cell Interactions
Cadherins are calcium-dependent transmembrane glycoproteins that are expressed in the early stages of development of various tissues. There are three primary cadherins: epithelial (E), neural (N), and placental (P) (67). Each, found commonly in desmosomes and cell junctions, activates intra- and intercellular communication (14). The cadherin intracellular portion connects, through α– and/or β-catenin, to actin microfilaments, anchoring cells to one another (2). Additionally, cadherins interact with various cell surface receptors, including receptor tyrosine kinases, such as epidermal growth factor receptor, to propagate intracellular signals resulting in migration, apoptosis, growth control, and differentiation of tumor cells (66).
E-cadherin, a metastasis suppressor molecule, is the most studied member of the cadherin family (68); its downregulation has been associated with transition to a mesenchymal phenotype, increased invasiveness, tumor progression, and poor survival (2,69,70). When E-cadherin expression is decreased, it is countered by upregulation of N- and P-cadherins. This cadherin switch also has been noted during normal development when migration, budding, and other invasive behaviors are necessary, such as during embryogenesis (71). Physiologic cadherin switching occurs when cells have reached their secondary site and the cadherins again switch and cells alter phenotype. This behavior, mimicked in tumor during epithelial–mesenchymal transition albeit without a secondary cadherin switch, leads to release of β-catenin and its translocation to the nucleus (3). β-catenin then activates signaling for proliferation and invasion (72).
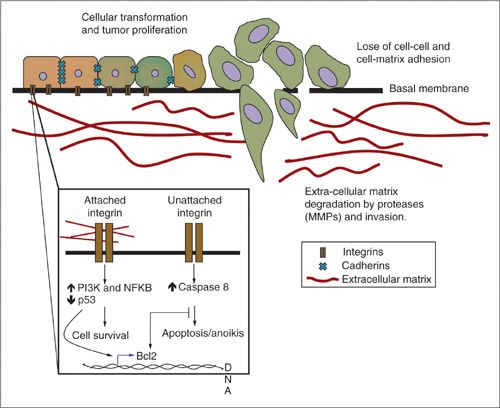
FIGURE 4.4. Invasion. Cancer cells lose their cell-to-cell (cadherin) and cell-to-matrix (integrin) attachment releasing single cells and allowing for cell migration. The release of proteases (MMPs, serine proteases) promotes ECM degradation forming tracks for cancer cells to migrate and invade surrounding tissues. Integrins can initiate prosurvival or proapoptotic signals depending on their attachment to the ECM. Metastatic cancer cells have acquired mechanisms to resist anoikis.
E-cadherin downregulation is associated with higher-grade, increased peritoneal seeding, and decreased overall survival in ovarian cancer (73). Sawada et al. demonstrated that when E-cadherin was silenced, it led to an increase adherence to mesothelial cells in an ovarian cancer in vivo model (74). E-cadherin expression has been shown to be lower in tumor cells floating in ascites, pleural effusions, and at the metastatic site, than in the primary tumor (2,75). Downregulation of E-cadherin results from upregulation of a series of zinc-finger transcriptional repressors, Snail, Slug, ZEB1/2, Twist, and SIP1, expression of which is also associated with increased invasive capability of ovarian cancer cells, resistance to chemotherapy, and poor clinical behavior in ovarian cancer (2,3,66,69,70). Therefore, the role of cadherins, particularly sustaining E-cadherin expression, is a critical defense against cancer cell invasion, migration, and adhesion by maintaining tight cell-to-cell connections.
Cell–Matrix Interactions
The ECM is a complex proteinaceous structure surrounding and supporting cells. It is composed of three major classes of biomolecules: proteoglycans, structural proteins such as collagens and elastins, and specialized matrix proteins such as fibronectin and laminins (76). The ECM can serve many functions, including cellular scaffolding support, binding site for growth factors and cofactors, and modulator of intercellular communication (76). ECM remodeling helps determine polarity and growth of tissues and has become a target of interest in cancer treatment. Many adhesion factors such as integrins, selectins, and CD44 interact not only with other cells, but also with the ECM.
Integrins These are transmembrane receptors named for their ability to integrate the microenvironment and cell signaling. They transmit signals bidirectionally, into and out of the cell, incorporating crosstalk with a variety of cell surface receptors and intracellular signaling proteins (66,77). Integrins are composed of eight α and eighteen β chains combining into 24 heterodimers each of which interacts with a specific repertoire of ligands (67). Each subunit contains a large extracellular domain, a single membrane-spanning domain, and a short cytoplasmic tail (78). Common integrin ligands include the major structural ECM molecules, thrombospondin, fibrinogen, and von Willebrand factor, among others (79). Cell-surface ligands for integrins include members of the immunoglobulin CAM superfamily, such as intercellular adhesion molecule-1 (ICAM-1) (79).
The tripeptide sequence, Asp–Gly–Arg (RGD), is the important ligand recognition site for integrins (80). Binding, whether through RGD or other recognition sites, causes integrin clustering or capping, β-subunit phosphorylation, and association of the cytoplasmic tail with cytoskeletal structures. The actin cytoskeleton is connected to the cytoplasmic integrin tail through the ERM proteins, ezrin, radixin, and moesin (81). Ligand binding also initiates intracellular signaling cascades that provide the machinery needed for cell motility and invasion, and to support survival (3,66,82). Although widely distributed throughout the body, integrins appear to be expressed in low and tightly organized quantities in normal tissue, and up- and dysregulated in tumors, including in gynecologic cancers (82). The α4β1, found commonly on mesothelial and endothelial cells, has been associated with increased peritoneal metastasis, and αvβ3, the vitronectin receptor, is both a driver of tumor proliferation and angiogenesis, via crosstalk with the VEGF receptor, in ovarian cancer (83,84). Integrins αvβ3 and αvβ6 have been associated with decreased patient survival in cervical cancer (85,86).
RGD and integrin binding has been used to focus drug and immunotherapy delivery in preclinical models (87,88). Hood et al. took advantage of integrin-facilitated drug delivery using αvβ3-targeted nanoparticles to deliver a mutant RAF1 gene into tumor vasculature (89). Xiao found that an integrin-targeting peptide, OA02, enhanced the efficiency of the uptake of nanoparticles into ovarian cancer cells (90). Paclitaxel was added to these nanoparticles and enhanced cytotoxicity was observed. This experiment was validated in ovarian tumor-bearing mice, where paclitaxel-loaded OA02-nanoparticles reduced tumor burden with lower systemic toxicity. Additional experiments have shown that integrin-linked doxorubicin-loaded nanoparticles delivered to tumor vasculature caused inhibition of metastasis and eliminated toxicity normally associated with systemic administration (62,77). Combining vascular targeting antagonists with chemotherapy also is hypothesized to cause more direct penetration of the drug into the tumor, while reducing systemic adverse events (77,78). This treatment concept is awaiting clinical validation for gynecologic cancers.
Selectins This class of molecules contains an epidermal growth factor-like motif and binds glycoproteins and glycolipids bearing sialyl Lewis X (sLex) in a calcium-dependent manner (80,91). They initiate the recruitment of lymphocytes by tethering and rolling these cells along the endothelium and are expressed predominantly on leukocytes, platelets, and endothelial cells (91). There are three types of selectins (66). Endothelial (E)-selectin, is triggered by cytokines such as TNFα and interleukin-1, and has a role in leukocyte adhesion at sites of inflammation and/or injury. Platelet (P) selectin is stored in endothelial cells and platelet granules and is activated by histamine and/or thrombin during inflammation. Lastly, leukocyte (L)-selectin is constitutively expressed on leukocytes and acts as a homing receptor for these cells to enter secondary lymphoid tissues. Elevated E-selectin has been associated with various diseases and cancers, including ovarian cancer. E7, an HPV-16 oncoprotein, upregulates the expression of E-selectin in cervical cancers (92). Selectins are thought to interact with tumor cells to shield them while in the circulation to enable subsequent extravasation and migration at the secondary site (66).
CD44, an E- and L-selectin ligand, has been correlated with cancer progression, metastasis, and poor overall survival (66,93). CD44 is the major hyaluronic acid receptor and is expressed by all nucleated cells (63,67). The interaction of CD44 expressed on ovarian cells with hyaluronic acid on mesothelial cells is thought to play a role in tumor dissemination and survival in ovarian cancer (3,55,94). Knockdown of CD44 led to a decrease in adhesion and invasion of ovarian cancer cells (63,94). It has been shown to upregulate proteases involved in breakdown of the ECM, a behavior required for invasion (95). Strobel et al. found that an antibody to CD44 inhibited peritoneal implants of ovarian cancer cells in a mouse model (96). Splice variants of CD44 have the ability to connect to the cytoskeleton, activate Src and Rho GTPase signal pathways, bind chemokines and growth factors, and to act as a coreceptor for other adhesion molecules (3,67).
Therefore, migration of cancer cells into or from blood to tissue requires coordinated sequential interactions of tumor cells with numerous adhesion molecules. The loss of E-cadherin leads to the loss of cell–cell junctions releasing single cells and facilitating migration away from the primary tumor and intravasation (97). Next, selectins facilitate tumor cell adhesion to vascular cells allowing them to transmigrate between the endothelium, followed by basement membrane dissolution and migration into the secondary site. Integrin-associated adhesion to basement membrane and ECM is involved in tumor cell exit from the primary tumor, entry into and exit from the vasculature, and attachment and growth at sites of metastasis. Although the basic concept of targeting these elements in therapy has been studied in animal models, translation to human disease has proved difficult to date.
INVASION/PROTEOLYSIS
The initial steps of invasion include activating signal pathways that regulate cell–cell junction breakdown, tumor or other cell-mediated ECM proteolysis, and migration away from the primary site. Local dissolution of the epithelial or endothelial basement membrane and/or ECM is the key event required for successful invasion. After migration from the primary and successful transit, tumor cells activate mechanisms that permit attachment to endothelial cells or endothelial cell basement membrane or local ECM at secondary sites. Success of this multistep process of invasion leads to successful metastasis.
Proteases are crucial for the degradation of the ECM, which places the tumor cells in direct contact with other cells and creates spaces or tracts through which tumor cells can migrate. Both tumor cells and local cells, such as endothelial cells, stromal, and inflammatory cells, can be the source of activated proteases (98). Proteolysis also unmasks ligands to which receptors of endothelial cells interact, such as integrin RGD moieties (20). The breakdown of the ECM also creates protein fragments, some of which promote and some that inhibit adhesion and migration functions; release of ECM-bound growth factors can also occur. Proteases that are secreted or attached to the cell surface can activate other proteases and cell surface receptors, multiplying the invasion stimulus and allowing adaptive changes in the microenvironment (3).
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are a family of metalloenzymes that require cleavage for activation. There are 5 categories: interstitial collagenases, gelatinases, stromelysins, membrane-type, and elastases (99,100). Each of these subclasses has selective substrate specificity with elements of the basement membrane and ECM constituting major substrates. MMPs are secreted by a variety of cancers with MMP-2, -7, and -9 described as active in the gynecological cancer microenvironment (101). The source of the MMP in the microenvironment remains a controversy, with data suggesting that both tumor and microenvironmental elements are involved in a cooperative fashion.
Greater quantity of activated MMP2 was observed by immunohistochemistry staining and immunoblots in ovarian cancer than in normal ovarian tissue (102). MMP2 expression was noted to be higher in stromal cells of invasive cervical cancer compared to CIN3. The increased expression was also correlated with increased stage and worse prognosis (103). A MMP7 polymorphism (MMP7-181G/G) has been demonstrated to be a potential risk factor for developing both endometrial and cervical cancers. It also is associated with an increased risk of higher grade and stage tumors and lymphatic metastasis (102, 104, 105). Overexpression of MMP7 has been shown in a variety of ovarian cancer cell lines and surgical specimens. Additionally, high levels of MMP9 have been found in ovarian xenograft models (106, 107). Elevation of MMPs is not specific to malignant tumors. Other benign tumors and conditions and normal remodeling behaviors may have increased quantities of MMPs (108). This may be one of the reasons that targeting MMPs was previously found to be unsuccessful due to inadequate doses and unexpected adverse events (109, 110). Therefore, the importance of this class of enzymes is recognized, although the therapeutic usefulness of targeting these elements is questionable.
Serine Proteases
Serine proteases are ubiquitous, in a variety of tissues, both constitutively and by induction. They have roles in wound healing and immune response, as well as promoting tumor growth, invasion, and metastasis (111). Plasminogen is an inactive proenzyme that is converted to the active enzyme plasmin, a key protease. Among the many functions of plasmin other than clotting is its requirement for propeptide cleavage and activation of MMPs (99). The plasminogen/plasmin system, in concert with the MMPs, helps regulate the tightly controlled process of ECM degradation. Additionally, this system plays a role in thrombolysis, embryogenesis, cell invasion, and angiogenesis (100).
Hepsin is a type II transmembrane serine protease initially shown to be overexpressed in prostate, renal, and ovarian cancers. Its message is upregulated in 90% of prostate cancers and appears to be exclusively expressed by tumor cells (112). More recently, Matsuo et al. found hepsin to have the strongest expression in endometrial cancer, compared with normal tissue and endometrial hyperplasia. They determined that strong hepsin expression was associated with decreased progression-free and overall survival, although not functioning as an independent prognostic factor (111). Ovarian tumors may also overexpress hepsin (113, 114). Xuan et al. showed that antihepsin antibodies significantly inhibited invasion of ovarian cancer cell lines (115). Hepsin may be underappreciated in gynecologic cancer biology.
Serine Protease Inhibitors
There is a large complex family of serine protease inhibitors, the SERPINS (116), some of which are upregulated and functional in gynecologic cancer progression (117, 118). One such serine protease inhibitor, secretory leukocyte protease inhibitor (SLPI), a whey acidic protein with anti-inflammatory properties, is overexpressed in breast and epithelial ovarian cancers. Paradoxically, SLPI has been shown to have a progrowth and proinvasion role in ovarian cancer, in part through activation of MMP9, and to be associated with a more aggressive tumor phenotype (119, 120). More recently, high levels of SLPI were shown to cause paclitaxel resistance in ovarian cancer cell lines (121). These data have credentialed this serine protease inhibitor as a potential molecular target in ovarian cancer.
The serine protease inhibitor maspin, SERPINB5, has been found to have an antiproliferative and antiangiogenic role in breast, lung, and prostate cancers. Its overexpression in ovarian cancer is associated with promotion of invasion and metastasis (122). Localization of maspin is the deciding factor leading to these opposing roles in cancer. Further studies are needed to clarify this issue.
Protease function constitutes a common requirement in any tissue remodeling process, ranging from embryogenesis to cancer. Hence, it was recognized early on that the metastatic potential of cancer cells relies on the ability to invade and degrade basement membranes (123). Cancer cells benefit from proteolysis to manage their attachment to and detachment from the ECM in addition to providing the overall structure of their invasion and interaction with the microenvironment. The ability to regulate matrix proteolysis and invasion is regarded to be essential in the treatment of cancer.
ANGIOGENESIS AND LYMPHANGIOGENESIS
Angiogenesis
Angiogenesis, the development of new blood vessels, has a key role in cancer progression and development of metastases. Its role in tumorigenesis was first described in 1971 by Folkman. He described that a tumor mass larger than 0.125 mm2 exceeds its capacity to acquire nutrients by simple diffusion, and that further expansion of the tumor would require new blood vessel formation (124–126). Not only do these new vessels provide growing tumors with the necessary nutrients, but they also serve as a route for metastatic dissemination (1). Angiogenesis is regulated by a balance of proangiogenic and angiostatic factors that result from the interaction between tumor cells, endothelial cells, and the microenvironment (127). Inhibitors of angiogenesis can help create a dynamic balance in the tumor milieu, from an angiogenic to a quiescent state, favoring the former one when hypoxia and other stresses are present.
Microvessel density is a method used to quantify vessels in cancer, yielding results that correlate with disease relapse and/or survival in cervical cancer (128), ovarian cancer (129–131), and endometrial cancer (132). The microvessel density significantly and independently correlated with progression free survival and overall survival in endometrial cancer (133). The antibody used to stain the microvessels can also give further information as to the quality of the vessels; CD105, staining endoglin, recognizes more immature vessels and CD31, anti-platelet/endothelial (PE)-CAM, recognizing microvessels (134).
New vessels develop in two ways, by sprouting or splitting. During sprouting, endothelial cells are activated by multiple growth factors and proteases are released, remodeling the surrounding ECM (Fig. 4.5) (135). This allows migration of activated endothelial cells through the matrix. Endothelial cell cords are formed, followed by polarization and formation of the lumen in the immature vessels (136,137
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


