and Fatemeh Ghane Sharbaf2
(1)
Department of Pediatrics Section of Nephrology, Rush University Medical Center, Chicago, IL, USA
(2)
Department of Pediatrics Section of Nephrology, Mashhad University of Medical Sciences, Mashhad, Iran
Introduction
Acute kidney injury (AKI) is a common problem in hospitalized patients and is associated with significant mortality and morbidity [1–5, 8–12, 14, 16, 17, 19–23, 25, 31, 32, 34, 39]. It occurs in 5 % of all hospitalized patients and up to 30 % of critically ill patients [1, 4, 8, 11, 18, 25, 30, 38]. The morbidity and mortality rates are 40 % and 60 %, respectively, in all patients admitted to the pediatric intensive care unit (ICU) [6, 14, 31, 32, 35, 36].
The preponderance of evidence supports the causal link between AKI and subsequent chronic kidney disease (CKD). Unlike a large proportion of adults who develop AKI superimposed on a substrate of hypertension, diabetes mellitus, chronic vascular disease, and underlying CKD, these factors generally do not underlie the development of AKI in children. Thus, the subsequent development of CKD in children surviving an episode of AKI is more readily attributable to residual kidney injury for the AKI.
Severe acute renal injury (AKI) necessitating renal replacement therapy often occurs in a significant proportion of critically ill patients, including patients with septic shock and multiorgan failure [21, 24, 27, 30, 31]. The critically ill and hemodynamic instable children with AKI are frequently treated with continuous renal replacement therapy (CRRT) [6, 7, 10, 13, 26, 27, 33]. CRRT, unlike the traditional hemodialysis and peritoneal dialysis, provides a slow and gentle fluid removal from body much like the native kidneys and removes inflammatory mediators of sepsis such as interleukin, TNF-alpha, and complement [6, 7, 10, 13, 24, 26, 27, 33, 37]. CRRT also provides adequate nutritional support for the catabolic AKI patients and for a controlled desired fluid balance [20].
Underdiagnosed and untreated AKI may lead to CKD and end-stage renal disease. The incidence of CKD and end-stage renal disease has nearly doubled over the last two decades across the globe. With annual mortality for patients on dialysis in the range of 20 %, more people die with treated uremia than with any cancer, except for lung cancer. If current trends continue, the toll of end-stage renal disease will exceed that of lung cancer. The burden of disease is paralleled by the enormous cost for delivering end-stage renal disease care [3, 14].
CRRT has become the preferred choice for blood purification and volume control in critically ill children [6, 7, 10, 13, 24, 26, 27, 33, 37]. The reported overall survival rate for children with severe AKI requiring CRRT is 60 % and mortality in infants is comparable with that of older children and adolescents [8, 15, 18, 22, 30].
CRRT treatments have undergone considerable evolution with the advent of new technologies over the last decade [28, 29].
CRRT is indicated in the pediatric population for acute renal failure (AKI), electrolyte abnormalities, catabolic patients with increased nutritional needs, patients with sepsis, poisoning (occasionally in combination with hemodialysis, inborn errors of metabolism, diuretic unresponsive hypervolemia, and hepatic or drug-induced coma) [6, 7, 13, 27, 34, 37]. Additionally, CRRT in conjunction with other therapies such as extracorporeal membranous oxygenation (ECMO), patients with cardiomyopathy on a left-ventricular assist device (LVAD), and the newer hepatic support therapies has also proven to be quite useful [9, 33]. A child with AKI and fluid overload with hemodynamic compromise is a common scenario that many of us would also consider a good candidate. The use of CRRT for inborn error of metabolism is less efficient then hemodialysis yet has been shown to be effective [6].
Several controversies, however, exist over which approach to patient care is most desirable and which form of CRRT should be utilized to achieve the best possible outcome. There are also controversies with respect to what should be the indications for starting such therapy, what is the preferred vascular access, and what kind of anticoagulation should be used. There is also uncertainty with respect to what membranes and filters are preferable and what machines are most appropriate for the patient needs.
While the basic principles of CRRT are similar for adults and children, the application of these modalities in children requires recognition of the unique properties of pediatric hemofiltration. Specific attention to detail, such as extracorporeal blood volume/blood priming (especially in patients <10 kg), nutritional issues, etiological differences in disease processes (i.e., inborn errors of metabolism), access, and line/membrane choice, must be given when dealing with problems in this population [6, 7, 10, 26–29, 37].
References of Section Introduction
1.
Andreoli SP. Acute renal failure. Curr Opin Pediatr. 2002;14:183–8.
2.
Andreoli SP. Acute renal failure in the newborn. Semin Perinatol. 2004;28(2):112–23.
3.
Assadi F. Strategies to reduce the incident of chronic kidney disease in children: time to change. J Nephrol. 2012;26:41–7.
4.
Bagshaw SM. Epidemiology of renal recovery after acute renal failure. Curr Opin Crit Care. 2006;12(6):544–50.
5.
Barletta GM, Bunchman TE. Acute renal failure in children and infants. Curr Opin Crit Care. 2004;10:499–504.
6.
Braun MC, Welch TR. Continuous venovenous hemodiafiltration in the treatment of acute hyperammonemia. Am J Nephrol. 1998;18(6):531–3.
7.
Bunchman TE, Maxvold NJ, Kershaw DB, et al. Continuous venovenous hemodiafiltration in infants and children. Pediatr Nephrol. 1994;8:96–9.
8.
Bunchman TE, McBryde KD, Mottes TE, et al. Pediatric acute renal failure: outcome by modality and disease. Pediatr Nephrol. 2001;16(12):1067–71.
9.
Chen H, Yu RG, Yin NN, Zhou JA. Combination of extracorporeal membrane oxygenation and continuous renal replacement therapy in critically ill patients: a systemic review. Crit Care. 2014;18(6):675.
10.
Flynn JT. Choice of dialysis modality for management of pediatric acute renal failure. Pediatr Nephrol. 2002;17:61–9.
11.
Garist C, Favia Z, Ricci Z, Averadi M, Picardo S, Cruz DN. Acute kidney injury in the pediatric population. Contrib Nephrol. 2010;165:345–56.
12.
Goldstein SL. Acute kidney injury in children: prevention, treatment, and rehabilitation. Contrib Nephrol. 2011:174:163–72.
13.
Goldstein SL. Overview of pediatric renal replacement therapy in acute renal failure. Artif Organs. 2003;27(9):781–5.
14.
Goldstein SL, Devarajan P. Acute kidney injury leads to pediatric patient mortality. Nat Rev Nephrol. 2010;6:393–4.
15.
Goldstein SL, Currier H, Graf JM, et al. Outcome in children receiving continuous hemofiltration. Pediatrics. 2001;107(6):1309–12.
16.
Hui-Stickle S, Brewer ED, Goldstein SL. Pediatric ARF epidemiology at a tertiary care center from 1999 to 2001. Am J Kidney Dis. 2005;45:96–101.
17.
Kaufman J, Dhakal M, Patel B, Hamburger R. Community-acquired acute renal failure. Am J Kidney Dis. 1991;17(2):191–8.
18.
Levy EM, Visconti CM, Horwitz RI. The effect of renal failure on mortality: a cohort analysis. JAMA. 1996:1489–94.
19.
Markowitz GS, Perazella MA. Drug-induced renal failure: a focus on tubulointerstitial disease. Clin Chim Acta. 2005;351(1–2):31–47.
20.
Maxvold NJ, Smoyer WE, Custer JR, Bunchman TE. Amino acid loss and nitrogen balance in critically ill children with acute renal failure: a comparison between CVVH and CVVHD therapies. Crit Care Med. 2000;28:1161–5.
21.
Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39(5):930–6.
22.
Nisula S, Kaukonen KM, Vaara ST, et al. Incidence, risk factors and 90-day mortality of patients with acute kidney injury in Finnish intensive care units: the FINNAKI study. Intensive Care Med. 2013;39:420–28.
23.
Norman ME, Assadi F. A prospective study of acute renal failure in the newborn infant. Pediatrics. 1979;63:475–79.
24.
Parekh RS, Bunchman TE. Dialysis support in the pediatric intensive care unit. Adv Ren Replace Ther. 1996;3:326–3.
25.
Perazella MA. Drug-induced nephropathy: an update. Expert Opin Drug Saf. 2005;4(4):689–706.
26.
Ronco C, Kellum JA, Mehta RL. Acute dialysis quality initiative (ADQI). Nephrol Dial Transplant. 2001;16(8):1555–8.
27.
Ronco C, Tetta C, Mariano F, et al. Interpreting the mechanisms of continuous renal replacement therapy in sepsis: the peak concentration hypothesis. Artif Organs. 2003;27(9):792–801.
28.
Ronco C, Ricci Z. Pediatric continuous renal replacement: 20 years later. Intensive Care Med. 2015;41(6):985–93.
29.
Ronco C, Ricci Z, Goldstein SL. Revolution in the management of acute kidney injury in newborns. Am J Kidney Dis. 2015;May 7. Pil:S0272(15)00620-4. doi:10.1053/J.ajkd.2015.03.029 [Epub ahead of print].
30.
Roy AK, Hoste EA, Clermont G, Kersten A, et al. RIFLE criteria for acute kidney injury is associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care. 2006;10:R73.
31.
Salahudeen AK, Doshi SM, Pawar T, Nowshad G, Lahoti A, Shah P. Incidence rate, clinical correlates, and outcomes of AKI in patients admitted to a comprehensive cancer center. Clin J Am Soc Nephrol. 2013;8:347–54.
32.
Schetz M, Dasta J, Goldstein S, Golper T. Drug-induced acute kidney injury. Curr Opin Crit Care. 2005;11(6):555–65.
33.
Tetta C, D’Intini V, Bellomo R, et al. Extracorporeal treatments in sepsis: are there new perspectives? Clin Nephrol. 2003;60(5):299–304.
34.
Wan L. Bellomo R, Di Giantomasso D, Ronco C. The pathogenesis of septic acute renal failure. Curr Opin Crit Care. 2003;9(6):496–502.
35.
Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:844–61.
36.
Wang HE, Muntner P, Chertow GM, Warnock DG. Acute kidney injury and mortality in hospitalized patients. Am J Nephrol. 2012;35:349–55.
37.
Warady B, Bunchman T. Dialysis therapy for children with acute renal failure: survey results. Pediatr Nephrol. 2000;15:11–3.
38.
Williams DM, Sreedhar SS, Mickell JJ, et al. Acute kidney failure: a pediatric experience over 20 years. Arch Pediatr Adolesc Med. 2002;156:893–90.
39.
Yang F. Zhang L, Wu H, Zou H, Du Y. Clinical analysis of cause, treatment and prognosis in acute kidney injury patients. PLoS. 2014;9(2):e85214. doi:10.1371/journal.pone.0085214.
Acute Kidney Injury
Definition
AKI (previously called acute renal failure) is characterized by abrupt onset of renal dysfunction manifesting as oliguria (urine output <0.5 mL/kg/h, for 6–12 h, a decrease in glomerular filtration rate (GFR) and the rise of serum creatinine) [3, 4, 14, 16, 26, 71]. The incidence of AKI in children appears to be increasing, and the etiology of AKI over the past decades has shifted from primary renal disease to multifactorial causes, particularly in hospitalized children [8, 13, 34, 42, 54, 70]. The history, physical examination, and laboratory studies, including urinalysis and radiographic studies, can establish the likely cause(s) of AKI. The prognosis of AKI is highly dependent on the underlying etiology of the AKI. Children who have suffered AKI from any cause are at risk for late development of kidney disease several years after the initial insult [20, 28, 29, 37, 71, 72].
Typically, in infants and small children, a minimal urine volume of approximately 1.0 mL/kg/h is required to excrete waste [16, 26, 43, 51]. Children with AKI due to hypoxic/ischemic insults, HUS, acute glomerulonephritis, and other causes are more likely to demonstrate oliguria or anuria (urine output less than 500 mL/24 h in older children or urine output less than 1 mL/kg/h in younger children and infants). Children with acute interstitial nephritis, nephrotoxic renal insults including aminoglycoside nephrotoxicity, and contrast nephropathy are more likely to have AKI with normal urine output. The morbidity and mortality rates of non-oliguric AKI are less than those of oliguric renal failure. While the precise incidence and causes of AKI in pediatric patients is unknown, recent studies suggest that the incidence of AKI in hospitalized children is increasing.
An important cause of AKI in hospitalized children is in the setting of post-cardiac surgery and in children undergoing stem cell transplantation. AKI in such children is frequently multifactorial, with ischemic/hypoxic injury and nephrotoxic insults being important contributors; the pathophysiology of hypoxic ischemic injury and nephrotoxic insults are described below [1, 14, 26, 28, 51, 68]. No epidemiology studies using an established definition of AKI have been conducted in pediatric patients. As described below, in pre-renal AKI the kidney is intrinsically normal, and renal function promptly returns to normal with restoration of adequate renal perfusion, while, in acute tubular necrosis (ATN), the kidney has sustained intrinsic injury, which requires repair and recovery before renal function returns to normal. Neonates with severe asphyxia had a higher incidence of AKI, while neonates with moderate asphyxia developed AKI less often.
In the urine output criteria, a urine output of less than 0.5 mL/kg/h for 8 h indicates the patient is at risk and should trigger the physician to start therapy [16].
This definition is, however, unreliable due to the lag time between the initiating injury and loss of kidney function, which may delay effective therapies that are limited to the early stage. Serum creatinine concentration does not increase until about 50 % of renal function is lost and also varies with age, muscle mass, medication, acid–base, and hydration status [8].
The RIFLE Criteria
In 2004, AKI Network and Kidney Disease Improving Global Outcome (KDIGO) introduced the risk, injury, failure, loss, and end-stage renal disease (RIFLE) classifications for the definition of AKI. RIFLE was modified for pediatric use (pRIFLE) (Table 1.1) [2, 52]. pRIFLE is a modification of the adult RIFLE classification and consists of three graded levels of injury (risk, injury, and failure) based upon the magnitude of change in estimated GFR (e.g., changes in serum creatinine) or urine output and two outcome measures, loss of kidney function and end-stage renal disease.
Table 1.1
Pediatric RIFLE criteria
GFR criteria | Urine output criteria | |
|---|---|---|
Risk | eCCl decrease >25 % | <0.5 mL/kg/h × 8 h |
Injury | eCCl decrease >50 % | <0.5 mL/kg/h × 16 h |
Failure | eCCl decrease >75 % | 0.3 mL/kg/h × 24 h or Anuria ×12 h |
Loss | Complete loss of renal function >4 weeks | Complete loss of renal function >4 weeks |
End-stage renal disease (ESRD) | ESRD >3 months | ESRD >3 months |
Small rises in serum creatinine levels of at least 0.3 mg/dL have been incorporated as the minimum requirements for the stage injury into the current definition of AKI.
The premise of the RIFLE criteria is to guide physicians on when to initiate therapy for the AKI patient. The first three letters R, I, and F allow the physician to assess patient AKI status and when to initiate therapy. Severity of AKI was graded from mild (RIFLE-R, “risk”) to severe (RIFLE-F, “failure”) based on changes in serum creatinine or estimated creatinine clearance and urine output. For example, a patient comes in with a serum creatinine of 1.0 mg/dL. A day later the patient’s creatinine rises to 1.5 mg/dL. This patient should be placed in the risk category and therapy should be initiated [2, 52].
The last two letters, L and E, are considered outcome categories, and this basically indicates that if therapy at R, I, or F is not initiated, the mortality rate increases, or the patients will become dialysis dependent [2, 52]. Patients in the F category exhibit a higher mortality rate than those who were in the R and I category. This would indicate that R and I should trigger early initiation, which enhances survival rate. These efforts to standardize AKI definition are a substantial advance, although areas of uncertainty remain [2, 52].
Despite significant improvements in clinical care, the mortality and morbidity associated with AIK remains high (>50 %) and can lead to end-stage renal disease [20, 28, 29, 51, 70, 72]. The lag time between the initiating renal insult and loss of renal function as assessed based on serum creatinine may explain the high mortality and morbidity rates associated with the AKI. It is therefore desirable to have an alternative AKI biomarker less influenced by such errors yet capable of detecting minor degrees of renal impairment in the sick patients. Novel biomarkers may further refine the definition of AKI, but their use will need to produce tangible improvements in outcomes and cost effectiveness [5, 7, 8, 31].
Determination of Glomerular Filtration Rate
AKI alters both the pharmacokinetics and pharmacodynamics of the prescribed drugs. As the GFR decreases, the elimination of drugs that are primarily excreted by the kidneys also decreases proportionally. Thus, assessment of renal function is important and should include measurement of GFR.
The renal clearance of inulin is the most reliable method for determination of a true GFR, but its use is limited because of expense, lack of availability, and problems of collecting timed urine samples in infants and children. GFR estimation (eGFR) from the renal clearance of creatinine has been widely used in the pediatric population [57–62, 66]. Creatinine-based eGFR without urine collection has been used frequently in children. There are several ways to estimate GFR in children. One of the easiest and more practical one is Schwartz formula, which takes and uses the concept of height as a measure of muscle mass divided by serum creatinine [57, 58].
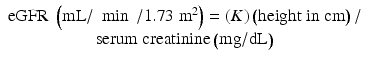 The value of K is 0.45 for term infants throughout the first year of life, 0.55 for children up to17 years and adolescent girls, and 0.7 for adolescent boys [58–60].
The value of K is 0.45 for term infants throughout the first year of life, 0.55 for children up to17 years and adolescent girls, and 0.7 for adolescent boys [58–60].
In children, there is not a single formula r ecommended for all ages. Schwartz equation can be used to estimate GFR in children and adolescents but should be used with caution or avoided in premature infants in the first several months of life.
However, the use of Schwartz equation carries several limitations. First, with changing renal function, the serum creatinine concentration will no longer reflect the true eGFR. As a result, eGFR using Schwartz equation requires that renal function should be stable and that the serum creatinine measurement is constant. Second, the use of eGFR by Schwartz formula in patients with nutritional compromise as is common in children with cancer undergoing chemotherapy may overestimate the GFR values with subsequent overdosing of nephrotoxic agent. Similarly, in patients with CKD, falsely elevated creatinine clearance can occur due to the increase in external creatinine excretion [9]. The addition of cimetidine to inhibit tubular creatinine secretion permits accurate measurement of creatinine clearance in patients with CKD.
Recently, Schwartz et al. adapted the traditional Schwartz equation to children and adolescents but did not find different k-coefficients between children and adolescents [42]. De Souza and colleagues used linear mixed-effect models to reestimate the 2009 Schwartz k-coefficient in 360 consecutive French subjects aged 1–18 years [24]. They assessed the agreement between the estimated GFR obtained with the new Schwartz formula and the rate measured by inulin clearance . In De Souza, k was estimated at 0.325 in boys <13 years and all girls and at 0.365 in boys aged ≥13 years. The performance of this formula was higher than that of 2009 Schwartz formula in children <13 years. This was first supported by a statistically significant reduction of the overestimation of the measured GFR in both cohorts, by better 10 and 30 % accuracies, and by a better concordance correlation coefficient. These investigators concluded that the performance and simplicity of Schwartz formula are strong arguments for its routine use in children and adolescents. The specific coefficient for children aged <13 years further improves this performance.
Overestimation of GFR using creatinine-based equations have resulted in higher than intended carboplatin doses with a potential for increased toxicity. This concern for patients’ safety led to the development of Calvert formula to determine the optimal carboplatin dosage [67]. Calvert formula is used to cap the maximum carboplatin dose based on target area under the curve (AUC). It is recommended that the maximum carboplatin dose should not exceed the target AUC (Table 1.2).
Table 1.2
Maximum AUC-based carboplatin dosage
Target AUC | Maximum carboplatin dose (mg) |
|---|---|
6 | 900 |
5 | 750 |
4 | 600 |
Calvert formula [67]: Total carboplatin dose (mg) = Target AUC (mg/mL/min) × creatinine clearance (mL/min) + 25.
Whereas the Schwartz formula is the widely used formula for the estimation of GFR in children, the Modification of Diet in Renal Disease (MDRD) and Cockcroft–Gault equations are the most popular estimate for adults but are inaccurate for patients less than 18 years old [25].
Radioisotopes such as 51Cr-EDTA plasma disappearance are used in plasma disappearance GFR determinations; however, these are not ideal for use in children, especially for repeated studies [48]. The plasma disappearance of iohexol is a better alternative than 51Cr-EDTA, because it is safe and not radioactive, easily measured, and freely excreted by glomerular filtration [48].
GFR estimating equations, based on serum concentrations of creatinine or cystatin C, are popular clinically and in research studies. To date, however, there is no dependable substitute for an accurately determined GFR, and iohexol plasma disappearance offers the best combination of safety, accuracy, and reproducible precision [60].
More recently, G. J. Schwartz and associates developed several new equations to estimate GFR in children using patient’s height, serum creatinine, cystatin C, and blood urea nitrogen (BUN), of which the best equation, with the highest accuracy and correlation and the narrowest 95 % limits of agreement, was [61].
![$$ \begin{array}{c}\mathrm{eGFR}=39.1\times {\left[\mathrm{height}\left({\mathrm{m}}^2\right)/\mathrm{S}\mathrm{C}\mathrm{r}\left(\mathrm{m}\mathrm{g}/\mathrm{dL}\right)\right]}^{0.516}\times {\left[1.8/\mathrm{cystatin}\;\mathrm{C}\left(\mathrm{m}\mathrm{g}/\mathrm{L}\right)\right]}^{0.294}\times \\ {}{\left[30/\mathrm{BUN}\left(\mathrm{m}\mathrm{g}/\mathrm{dL}\right)\right]}^{0.169}\times \left[1.099\kern0.24em \mathrm{male}\right]\times {\left[\mathrm{height}\;\left(\mathrm{m}/1.4\right)\right]}^{0.188}\end{array} $$](/wp-content/uploads/2016/07/A370551_1_En_1_Chapter_Equb.gif) These authors also described a simple bedside equation of 0.413 × height (cm)/serum creatinine (mg/dL), which provides a good approximation of eGFR in children and adolescents with CKD based on serum creatinine measurements referenced to the iohexol-based GFR determinations [61].
These authors also described a simple bedside equation of 0.413 × height (cm)/serum creatinine (mg/dL), which provides a good approximation of eGFR in children and adolescents with CKD based on serum creatinine measurements referenced to the iohexol-based GFR determinations [61].
Urinary AKI Biomarkers
The incidence of both AKI and CKD is rising and reaching epidemic proportions [10]. In both situations, early intervention can significantly improve the dismal prognosis. Current standard assessments of renal function for pediatric patients use serum creatinine or formulas based on serum creatinine designed for longitudinal assessment of baseline renal function. It is accepted that the concentration of serum creatinine is an insensitive and delayed measure of decreased kidney function following AKI.
The accurate diagnosis of AKI is especially problematic in critically ill patients, in whom renal function is in an unsteady state, thus rendering such creatinine-based baseline assessment measures of renal function potentially inadequate.
Recently, a number of novel urinary AKI biomarkers including kidney injury molecule 1 (KIM-1), cystatin C (Cys-C), neutrophil gelatinase-associated lipocalin (NGAL), interleukin-18 (IL-18), and liver fatty acid-binding protein (L-FABP) have been identified and proved effective in predicting AKI prior to a change in serum creatinine levels [3–8, 13].
It is likely that they will be useful for timing the initial insult and assessing the duration and severity of disease (analogous to the cardiac panel for evaluating chest pain). Biomarkers may also serve to discern disease subtypes, identify etiologies, predict clinical outcomes, allow for risk stratification and prognostication, and monitor the response to interventions. The widespread availability of such information promises to revolutionize renal care in both children and adults and allow for the practice of personalized and predictive medicine at an unprecedented level.
Ischemic events, nephrotoxic drugs, and bacterial endotoxins are the frequent causes of AKI (about 90 %) [3, 4, 14, 16, 26, 28, 51, 68, 71, 72]. In these conditions, renal tubular injury is mediated through the release of pro-inflammatory cytokines and vasoactive substances leading to the necrosis of renal tubular epithelial cells [1]. ATN is followed by accumulation of biomarkers in the plasma and urine due to an increased synthesis in tubular cells (IL-18, KIM-1, NGAL) and/or impaired reabsorption in the proximal tubule (NGAL, cystatin C) [5, 7, 8, 31].
NGAL and IL-18 are pro-inflammatory cytokines that are produced and catabolized in the proximal tubule and have been reported as reliable markers of impaired proximal renal tubular dysfunction following ischemic and toxic injury, and after cardiac surgery, KIM-1 is known to mediate ischemic renal injury through activation of apoptosis in proximal renal tubular epithelial cells. Urinary KIM-1 appears to be more specific to ischemic or nephrotoxic injury and not significantly affected by prenatal azotemia, urinary tract infection, or CKD. Cys-C is a low-molecular-weight protein that is produced by all nucleated cells and freely filtered by glomerular filtration and then completely reabsorbed and catabolized by the healthy proximal tubular cells. Urine CYs-C levels increase when the proximal tubular cells are damaged. Sepsis and proteinuria may cause false-negative results [5, 7, 8, 31].
Of these biomarkers, NGAL, L-18, and KIM-1 have the highest potential for early diagnosis of AKI, and it is also recommended that a panel of biomarkers, rather than a single biomarker, will be needed to perform extremely well for mortality risk prediction after AKI [5, 7, 8, 31].
In children who underwent cardiac surgery, urinary NGAL concentration was highly predictive of subsequent clinical diagnosis of AKI (sensitivity 1.0, specificity 0.98), receiver operator characteristics (ROC) AUC = 0.99 at 2 h after surgery. The AUC for urinary IL-18 ranges up to 0.9 for the early detection of AKI with low sensitivity but high specificity (0.85–0.94). Urinary KIM-1 level had an AUC of 0.80 for the early detection of ischemic urinary. KIM-1 concentrations were elevated to a much higher degree in patients with ischemic AKI than in patients with other forms of AKI with an AUC of 0.80.
Premature infants have higher levels of urinary AKI biomarkers compared with term infants because of the underdeveloped kidney function [3–5]. In recent studies, Askenazi et al. [2, 8] found that the baseline values of urine NGAL, KIM-1, and Cys-C but not IL-18 decreased with increasing gestational age. These authors found that the baseline urine NGAL, KIM-1, and Cys-C concentrations in premature infants (≤26 weeks’ gestation) were 351 (ng/mL), 226 (pg/mL), and 911 (ng/mL), respectively. These values fell significantly to 85 (ng/mL), 143 (pg/mL), and 133 (ng/mL) for NGAL, KIM-1, and Cys-C, respectively, around 30 weeks of gestation. The mean urinary L-18 did not differ across gestational age categories and were 42 (pg/mL) for infants ≤26 weeks’ gestation and 67 (pg/mL) ≥30 weeks’ gestation, respectively. After the neonatal period, the median reference values for urine NGAL, KIM-1, and Cys-C are the following: 6.6 ng/mL, range 2.8–17; 410 pg/mL, range 226–703; and 20 ng/mL, range 0.5–200, respectively.
AKI Pathogenesis
Pre-renal azotemia and ischemic AKI are part of spectrum manifestations of renal hypoperfusion and can complicate any disease that reduces the “effective arterial blood volume” (e.g., cardiac failure, severe ischemia, hypovolemia) [1, 67]. Autoregulation of renal blood flow and GFR are overwhelmed at mean arterial blood pressure below 60–80 mmHg and AKI ensues. GFR may be impaired at lesser degrees of hypotension in neonates and those with preexisting renal diseases. Several drugs also perturb adaptive responses and can precipitate or aggravate pre-renal azotemia in subjects with renal hypoperfusion. These include nonsteroidal anti-inflammatory drugs (NSAIDs) and angiotensin-converting enzyme (ACE) inhibitors, which block biosynthesis of vasodilator prostaglandins and angiotensin II, respectively [38, 44, 55].
Patient-Related Risk Factors
Drug-induced renal disorders are more common in certain patients and in specific clinical situations. Infants and young children with extracellular volume depletion, sepsis, renal impairment, cardiovascular disease, diabetes, or prior exposure to radiocontrast agents are at risk of developing drug nephrotoxicity [72].
Drugs can cause AKI, intrarenal obstruction, interstitial nephritis, nephrotic syndrome, and acid–base and fluid–electrolyte disorders. Certain drugs can cause alteration in intraglomerular hemodynamics; inflammatory changes in renal tubular cells, leading to AKI; tubulointerstitial disease; and renal scarring. Drug-induced nephrotoxicity tends to occur more frequently in patients with intravascular volume depletion, diabetes, congestive heart failure, CKD, and sepsis [72]. Thus, early detection and treatment of drugs’ adverse effects are important to prevent progression to end-stage renal disease. Table 1.3 provides list of the commonly used nephrotoxic drugs.
Table 1.3
The most commonly used nephrotoxic drugsa
Pre-renal azotemia | Acute tubular necrosis | Acute/chronic interstitial nephritis | Tubular obstruction | Hypersensitivity angiitis | Thrombotic microangiopathy |
|---|---|---|---|---|---|
ACE inhibitors AT2-receptor antagonists Cocaine Cyclosporine Diuretics Estrogen Quinine Interleukins Mitomycin C NSADIs Tacrolimus | ACE inhibitors Acetaminophen Aminoglycosides Amitriptyline Amphotericin B AT2-receptor antagonists Cephalosporins Cisplatinum Contrast media Cyclosporine Dextran Diphenhydramine Furosemide Haloperidol Mannitol Methadone NSAIDs Heavy metals Pentamidine Tacrolimus Tetracycline | Acetaminophen Acetazolamide Acetylsalicylic acid Acyclovir Allopurinol Ampicillin Cephalosporins Chlorpropamide Ciprofloxacin Cimetidine Cisplatin Contrast media Fenoprofen Furosemide Indinavir Lansoprazole Lithium Methicillin Naproxen NSAIDs Omeprazole Oxacillin Pantoprazole Phenacetin Phenindione Phenytoin Phenylbutazone Ranitidine Rifampicin Sulfonamides Thiazides Vancomycin | Acyclovir Ethylene glycol Ganciclovir Methotrexate Probenecid Protease inhibitors Quinolones Sulfonamides Triamterene | Ampicillin Penicillin G Sulfonamides | Clopidogrel Cyclosporine Oral contraceptives Quinine Mitomycin C |
AKI Etiology
AKI has a long differential diagnosis. History can help to classify the pathophysiology of AKI as pre-renal, intrinsic renal, or post-renal failure, and it may suggest some specific etiologies.
Pre-renal Failure
Patients commonly present with symptoms related to hypovolemia, including thirst, decreased urine output, dizziness, and orthostatic hypotension. Ask about volume loss from vomiting, diarrhea, sweating, polyuria, or hemorrhage. Patients with advanced cardiac failure leading to depressed renal perfusion may present with orthopnea and paroxysmal nocturnal dyspnea.
Elders with vague mental status change are commonly found to have pre-renal or normotensive ischemic AKI. Insensible fluid losses can result in severe hypovolemia in patients with restricted fluid access and should be suspected in elderly patients and in comatose or sedated patients.
Pre-renal azotemia and ischemic AKI are part of spectrum manifestations of renal hypoperfusion and can complicate any disease that reduces the “effective arterial blood volume” (e.g., cardiac failure, severe systemic vasodilatation, hypovolemia) [1, 25]. Autoregulation of renal blood flow and GFR is overwhelmed at mean arterial blood pressure below 60–80 mmHg and AKI ensues. GFR may be impaired at lesser degrees of hypotension in neonates and those with preexisting renal diseases.
Pre-renal AKI represents the most common form of kidney injury and often leads to intrinsic AKI if it is not promptly corrected. Volume loss and decreased effective arterial blood volume can provoke this syndrome; the source of the loss may be gastrointestinal, renal, or skin (e.g., burns) or from internal or external hemorrhage. Pre-renal AKI can also result from decreased renal perfusion in patients with heart failure or shock (e.g., sepsis, anaphylaxis).
Several classes of medications can induce pre-renal AKI in volume-depleted states, including ACE inhibitors, angiotensin receptor blockers (ARBs), aminoglycosides, amphotericin B, and radiologic contrast agents. Arteriolar vasoconstriction leading to pre-renal AKI can occur with the use of radiocontrast agents, NSAIDs, amphotericin, calcineurin inhibitors, norepinephrine, and other vasopressor agents.
The patient’s age has significant implications for the differential diagnosis of AKI.
The most frequent cause of AKI in children and adolescents is under-perfusion of the normal kidney because of perinatal asphyxia. Various causes of hypovolemia and shock in the newborn may also result in AKI [1, 3, 4, 14].
To summarize, AKI can be caused by the following conditions:




Extracellular Volume Depletion
Renal losses – diuretics, polyuria
GI losses – vomiting, diarrhea
Cutaneous losses – burns, Stevens–Johnson syndrome
Hemorrhage
Decreased Cardiac Output
Heart failure
Pulmonary embolus
Abdominal compartment syndrome – tense ascites< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
