Fig. 13.1
Bone modeling and remodeling. Bone modeling occurs uniquely in children and results from the combined activities of osteoblasts, osteoclasts and the growth plate cells. Its result is bone that grows in length, width, and is reshaped (striped arrows). Compared to remodeling, it is a fast process in which all bone surfaces are active and osteoblasts and osteoclasts work at the same time. In contrast, bone remodeling takes place in both adults and children. It can occur in either trabecular or cortical bone as a consequence of microfractures, mechanical stress or triggered to replace old bone. Small amounts of bone are dissolved by osteoclasts, which are followed by a wave of bone-forming osteoblasts. The protein matrix released by osteoblasts then becomes calcified, restoring the original bone mass
Growth, Bone Modeling, and Remodeling
Childhood is a time of skeletal growth and maturation. After rapid growth in the third trimester of gestation and in the early neonatal period, bone growth rates fall sharply until puberty. Sexual maturation is associated with a dramatic acceleration in longitudinal bone growth until it ceases when growth plates become fused. The structure of bone also changes, with expansion of the medullary cavity and thickening of the cortical shell and of existing bone trabeculae. Consequently, the mechanical properties of bone evolve rapidly during adolescence. Bone mineralization lags behind linear growth, resulting in a relative structural weakness that increases fracture risk during adolescence [6, 7]. Peak bone mass is achieved after a period of consolidation at the end of the second decade of life in females and at the beginning of the third decade of life in males [8]. Consequently, gains in skeletal mass may occur after epiphyseal closure and cessation of linear growth. After a period of stability that lasts for about two decades, bone loss occurs after menopause in women and in elderly men. In adults, loss of mineral mass is accompanied by deterioration of bone microarchitecture and increased propensity to fractures with age, which defines osteoporosis [9]. This process of bone deterioration may be enhanced by IBD in adults, but the effects of the disease in children are probably different due to the uniqueness of the skeletal biology in individuals that are still growing. Although the total amount of bone tissue grows with the individual, the material density of bone (e.g., in g/cm3) remains relatively constant in children until the latter stages of sexual maturation, when it rises modestly [10, 11].
Bone mass is maintained by bone remodeling, characterized by the formation of a functional unit that consists of osteoclasts and osteoblasts (the bone remodeling unit) (Fig. 13.1). In response to damage or mechanical strain, osteoclasts resorb bone and form resorption pits. This is followed by recruitment of osteoblasts that fill in bone divots with a specialized protein matrix (osteoid, composed primarily of type I collagen). Osteoid later becomes relatively stiff by deposition of hydroxyapatite [1]. Some osteoblasts undergo apoptosis, while others become embedded in the newly formed bone matrix and become osteocytes. Osteocytes become interconnected by dendritic processes, sense mechanical strain and direct the cells of the bone remodeling unit [12]. The process of remodeling typically takes several months, and generates quantum amounts of bone. In bone remodeling, the activities of osteoblasts and osteoclasts are sequentially coordinated, so formation normally follows resorption. Only about 20% of bone surfaces in the body are actively engaged in this process at any given time. Bone remodeling occurs both in adults and children, and can take place in cortical or trabecular bone [13]. However, gains in bone mass in childhood are largely due a combination of the action of the growth plate and a distinct process called bone modeling, which is exclusive to children (Fig. 13.1). Bone modeling can be compared to the process of erecting a skyscraper, while bone remodeling is akin to maintaining the building’s structural integrity over time by scheduled and unscheduled (prompted by damage) repairs. In children, accumulation of bone mass is largely a consequence of linear growth and bone modeling [11]. Longitudinal growth is triggered by hormonal signals and occurs due to the production of a cartilaginous scaffold by the growth plate that is calcified, remodeled, and turned into trabecular bone. Trabeculae act as struts, columns and joists to distribute mechanical load from the epiphysis to the compact bone shaft. Linear growth and bone modeling occur simultaneously, with osteoblasts laying down new bone matrix in the periosteal surface, while osteoclasts reshape the bone by resorbing endosteal and metaphyseal bone (resulting in the expansion of the medullary cavity and metaphyseal inwaisting, respectively). Bone modeling occurs in 100% of bone surfaces, and both osteoclasts and osteoblasts are active at the same time, and is faster than bone remodeling [14].
These significant physiological differences between pediatric and adult bone have important implications for children with IBD. Disease and treatment factors influence both modeling and remodeling, but the major impact in children is likely to be on growth plate cells and bone formation, the two most active processes in bone during growth. Moreover, IBD is associated with significant deficits in muscle mass, leading to a decrease in mechanical strain on bone and a consequent reduction in bone modeling, and weight loss associated with IBD decreases gravitational forces on the skeleton.
The measurement of true volumetric bone density is not routinely available clinically. Instead, dual X-ray absorptiometry (DXA), a precise and accurate method commonly used to assess bone mass measures bone mineral content, which is divided by bone area (e.g., DXA “density” is expressed as g/cm2, or “areal” bone density, not a true material density). DXA produces a two-dimensional projection of three-dimensional bones, so larger bones with equal material density than smaller bones will be measured as “denser” DXA. Therefore, diseases like IBD that can affect linear growth and bone size may affect DXA measurements by underestimating bone mass in smaller children. This requires correction of DXA readings for patient’s size, sex, and sexual maturation [15] (please see chap. 21 by M. Thayu, E. Semeao, and M.B. Loenard).
Bone Cells and Inflammation
Osteoclasts and the RANKL/OPG System
Both bone remodeling and modeling involve the activity of osteoclasts and osteoblasts. Osteoclasts are cells from the macrophage/monocyte lineage, which secrete and are regulated by cytokines [16]. Osteoclasts are formed primarily by stimulation of hematopoietic precursors with receptor activator of nuclear factor κB-ligand (RANKL) in the presence of macrophage stimulating factor (M-CSF) (Fig. 13.2). RANKL, a member of the tumor necrosis factor receptor superfamily is produced by osteoblasts, stromal cells, fibroblasts and activated T cells [17, 18], and stimulates osteoclast differentiation, activation, and survival. RANKL-deficient mice have hyperdense bones secondary to lack of osteoclasts [17]. RANKL has been implicated as a key factor in the pathogenesis of bone loss associated with increased resorption, including postmenopausal osteoporosis [19] and rheumatoid arthritis [20]. Multiple inflammatory cytokines (tumor necrosis factor-α, interleukin 1-β) work via RANKL to increase osteoclast formation [21–23]. TNF-α can also modestly stimulate osteoclastogenesis directly [24, 25], and TNF−/− mice and mice lacking the p55 TNF receptor are resistant to the bone effects of estrogen deficiency in mice [26], suggesting an important role for this cytokine in bone loss due to increased bone resorption.
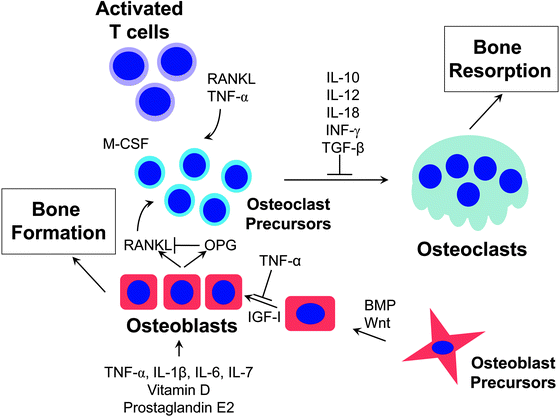
Fig. 13.2
Regulation of bone cells by cytokines. Both osteoblasts and osteoclasts are tightly regulated by a host of systemic and local factors. For osteoclasts, the “gatekeeper” regulator is RANKL. Cytokines and other factors influence osteoclast differentiation and activity by interacting with the RANKL pathway. Cytokines can either stimulate or inhibit osteoclastogenesis. RANKL is secreted by osteoblasts, stromal cells and activated T cells. Osteoblasts also release OPG, which neutralizes RANKL before it can bind to RANK on the surface of osteoclast precursors. Osteoblast formation is induced by IGF-1, bone morphogenetic proteins, Wnt and other growth factors. Inflammatory cytokines such as TNF-α can inhibit osteoblast formation by multiple mechanisms. Therefore, inflammation can affect both osteoblast and osteoclast function
Osteoprotegerin (OPG) is a soluble decoy receptor for RANKL produced by osteoblasts and stromal cells [27]. OPG serves as an inhibitor of osteoclast development. OPG transgenic mice have hyperdense bones, a phenotype that can be replicated by systemic administration of OPG to normal mice. OPG-null mice on the other hand are profoundly osteopenic due to unopposed osteoclast activity [28, 29]. Consequently, OPG is a key regulator of bone resorption. Besides OPG, another control switch in osteoclast development is interferon (IFN)-β, which is induced by RANKL binding to its receptor RANK on osteoclast precursors [30]. IFN-β interferes with the activity of c-fos, a transcription factor that is essential for osteoclast formation. Other factors, such as TGF-β and Wnt inhibit osteoclastogenesis by upregulating the production of OPG by osteoblasts [31, 32]. Wnt also directly represses RANKL expression [33]. In addition, several cytokines relevant to the pathogenesis of IBD inhibit osteoclast differentiation, including IFN-γ [34], IL-10 [35, 36], IL-12 [37], and IL-17 [38]. Moreover, osteoclast differentiation involves transcription factors such as NFκВ, AP-1, and NFATc1, as well as co-stimulation via immunoglobulin-like receptors and activation of the phosphatase calcineurin, which can be regulated by inflammation [39, 40]. Therefore, osteoclast formation is subject to multiple regulatory controls by cytokines, transcription factors and enzymes that play key roles in IBD. These pathways are an example of the close physiological ties between the immune system and bone cells. However, it is not yet known whether these mechanisms are engaged in regulating bone mass in children with IBD.
The RANKL/OPG system also plays important roles outside of bone. This is evidenced by the lack of peripheral lymph nodes and impaired development of lactating mammary glands in RANKL or RANK-null mice [41]. In addition, RANKL/OPG may be involved in the formation of calcified atherosclerotic plaques and serum OPG is emerging as a marker of cardiovascular mortality [42]. The balance between RANKL/OPG may affect the severity of bone metastases of several cancers [43]. RANKL contributes to normal dendritic cell function and survival, and the early development of B and T cells [17, 44]. In addition, RANKL/RANK may play a role in intestinal mucosal tolerance [45]. OPG also has a role in the regulation of the immune response. Both B cells and dendritic cells secrete OPG, and this secretion is regulated by the CD40 receptor [46]. Also, dendritic cells isolated from OPG−/− mice more efficiently present antigen in vitro and secrete more inflammatory cytokines when stimulated with bacterial products or soluble RANKL in vitro [46]. Collectively, this evidence suggests that RANKL/RANK/OPG play an important role in the regulation of the immune response, and in pathways involving the mobilization of calcium [47].
A role for the RANKL/OPG system is emerging in IBD. Circulating OPG levels are elevated in patients with IBD [3, 48, 49], and expression of OPG and RANKL is increased in colonic macrophages, dendritic cells and epithelial cells [49–51]. High fecal OPG (which probably comes from the inflamed colonic epithelium) predicts resistance to corticosteroids and to infliximab in patients with IBD [52, 53]. In addition, fecal OPG decreases in children with IBD in remission [54]. Currently it is not clear whether circulating OPG in patients with IBD represents spillover from intestinal inflammatory activity, or it comes from bone or other tissues (e.g., the endothelium), or both. The function of RANKL/OPG in the pathogenesis of intestinal inflammation deserves further study.
Osteoblasts
Osteoblasts are cells from mesenchymal origin. Several factors and hormones regulate osteoblast formation [55] (Fig. 13.2). Insulin-like growth factor-1 (IGF-1), is secreted by the liver in response to stimulation by growth hormone, and enhances the expression of the mature osteoblast phenotype [56]. Serum IGF-1 is frequently reduced in children with active IBD due to growth hormone insensitivity in the liver and malnutrition [57]. Consequently, relative IGF-1 deficiency in children with IBD may negatively affect osteoblast differentiation and function. TNF-α, an important cytokine in the pathogenesis of IBD inhibits osteoblast development by inducing the degradation of Runx2, a critical transcription factor in osteoblast development [58] and suppression of Wnt signaling [59]. TNF-α also regulates a number of inflammatory chemokines and cytokines, inflammatory genes, transcriptional regulators, bone-remodeling genes, signal transducers, cytoskeletal genes, and genes involved in apoptosis in pre-osteoblasts [60]. TNF-α and colitis decrease the expression of Phex in osteoblasts, which affects their mineralization function [61, 62]. In children, TNF-α blockade leads to a brisk increase in biomarkers of bone formation and significant linear growth, suggesting an activation of bone modeling [63]. However, the effects of infliximab may be a product of improved disease control and not specific effects of this drug on bone metabolism in these patients.
T Cells and Bone Loss
T cells are emerging as important regulators of bone cell function [64]. Activated T cells can regulate osteoclast formation and activity by several mechanisms, both RANKL-dependent and independent. Activated T cells secrete RANKL, and consequently can promote osteoclast differentiation and survival. Both soluble and membrane-bound RANKL is produced by activated CD4+ and CD8+ T cells [18]. T cell-induced bone resorption has been implicated in tissue injury in animal models of arthritis and periodontal disease [20]. T cells may also play an important role in bone loss associated with estrogen deficiency, where osteoclast activity is upregulated. This is suggested by experiments performed in ovariectomized mice, where absence of T cells prevents bone loss [22]. In this model, the expansion of a TNF-α producing T cell pool appears to be essential and may occur as a result of upregulation of antigen presentation. The nature of the activating antigen(s) is not yet known, but it is possible that both self and foreign epitopes (including intestinal bacterial products) may play a role [64]. The concept that T cells activated by bacterial antigens may regulate bone cell function is intriguing. In IBD, it is possible that activated T cells may serve as “inflammatory shuttles” between intestine and bone, since circulating T cells produce cytokines that can regulate both osteoblasts and osteoclasts. In addition, it is possible that circulating antigens may trigger immune responses in the bone marrow that affect bone cell function. These possibilities await additional research.
Effects of Intestinal Inflammation on Bone
Animal Models
IBD is a complex clinical entity, where multiple disease and treatment factors contribute to affect bone cell biology and ultimately skeletal health. In an effort to study mechanistic questions, animal models of intestinal inflammation have been used by several groups. A brief description of their observations follows.
Studies in both rat and mouse models suggest that intestinal inflammation can decrease bone mass by impairing bone formation. Lin et al. induced colitis in rats by rectal instillation of TNBS [65] to study its effects on bone mass, assessed by quantitative histomorphometry. After 3 weeks rats with colitis had a 33% loss of trabecular bone loss in the tibia compared with age-matched, pair-fed control animals. This was associated with a marked suppression of the trabecular bone formation rate. As the colitis healed, bone formation became more active and bone mass normalized after 12 weeks. In IL-10−/− mice with colitis Dresner-Pollak et al. performed bone densitometry, ash weight, histomorphometry analysis and mechanical fragility testing [66]. They observed that bone mass decreased secondary to decreased bone formation in 8 and 12 week old mice; bone resorption was not increased in mice with colitis compared to wild type controls. Long bones were more fragile in IL-10−/− with colitis, an ash weight was reduced. Harris et al. have demonstrated that the inhibition of bone formation and bone modeling is reversible with healing of colitis in mice [67].
Two reports using adoptive transfer models of colitis suggest that bone mass decreases secondary to increased bone resorption. In the first paper, Ashcroft et al. studied IL-2−/− mice with colitis at 4, 7, and 9 weeks of age, and compared X-ray, histomorphometry with IL-2−/+ and wild type mice. IL-2−/− mice develop colitis, but also have splenomegaly, anemia and other signs of systemic inflammation [68]. They observed a decrease in trabecular bone volume in IL-2−/− with colitis compared with the other two groups of mice at 7 and 9 weeks of age. C57BL/6-Rag1−/− mice transplanted with CD3+ cells from IL-2−/− had significantly lower femoral BMD and % trabecular volume 6–8 weeks post-grafting. Serum OPG and osteoclast number were significantly higher in mice engrafted with T cells from IL-2−/− mice compared to IL-2+/+. In this model, treatment with OPG was associated with both improved bone mass and decreased intestinal inflammation. These results point to a possible role of T cells in bone loss in the context of intestinal inflammation, and suggest a possible anti-inflammatory role for OPG. In the second study, Byrne et al. transferred CD4+CD45RBHi or CD4+CD45RBLo from CB6F1 mice to C.B.17 scid/scid mice [69]. CD4+CD45RBHi, but not CD4+CD45RBLo caused colitis in recipient mice, and mice with colitis had lower bone mineral density in the femur/tibia. To treat bone loss, mice received Fc-OPG 3.4–5 mg/kg SC three times weekly for 34 days. OPG had no effect on the severity of colitis, but significantly improved BMD.
Collectively, these observations suggest that intestinal inflammation can directly affect bone mass in rodents. Mechanisms may include decreased bone formation or increased bone resorption, depending on the model. It appears that intestinal inflammation present at an early age is associated with decreased bone formation. Administration of exogenous OPG increases bone mass in older mice with certain forms of colitis. However, this may be a non-specific effect of OPG on normally active osteoclasts and by itself does not establish that increased bone resorption is responsible for bone loss in rodent models of colitis. In addition, in adoptive transfer models of colitis, it is not possible to distinguish whether intestinal inflammation in the recipient animals caused bone loss or if colitogenic T cells directly migrated into the bone marrow influenced bone cell function. However, it is interesting that in the CD4+CD45RBHi model there is an inflammatory infiltrate in the bone marrow containing TNF-α-producing cells [69]. This provides proof of principle that intestinal inflammation is associated with the presence of activated T cells in the bone marrow that secrete pro-inflammatory cytokines which may influence the function of bone cells. The presence of these cells awaits studies in other animal models of intestinal inflammation and in humans.
Human Studies
Several studies have measured bone mineral density in children with IBD, both in incident and in prevalent cohorts (Table 13.1). Most of these studies are cross-sectional, and used DXA to measure BMD. These studies suggest that decreased bone mineral density is common in children with Crohn disease at the time of diagnosis, especially in those patients with delays in growth and sexual maturation, and those with decreased lean tissue mass [3, 70, 71]. These studies were performed in incident cohorts of treatment-naïve patients, and suggest that disease factors can affect bone mass in children with IBD prior to the initiation of treatment. Collectively, they suggest that children with Crohn disease are at greater risk for decreased bone mass than children with ulcerative colitis, perhaps because Crohn disease is more likely to affect linear growth than ulcerative colitis. Patients with low body mass index, low serum albumin and active severe IBD appear to be at particular risk for decreased BMD. According to recent guidelines by the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition, children with a new diagnosis of IBD should have DXA scanning [72]. In addition to measuring bone mass, body composition data provided by the instrument may be helpful in guiding the nutritional rehabilitation of these patients. It is important to DXA BMD measurements to patient size, gender, and sexual maturation, because in any given patient with IBD, the challenge is to distinguish between small, normally mineralized bones and abnormally thin and weak bones [72]. Taken together, these studies indicate that the observed reduction of BMD in children with IBD can be attributed in part to decreased bone size. However, it is important to note that smaller bones may be weaker, and their physical properties may not be normal. It is not yet known whether smaller bone size leads to increased fracture risk in children with IBD.




Table 13.1
Studies on bone density in children with IBD
Cohort | n | Age (years) | M/F | Normative data | Z-score | Risk factors | References | |
|---|---|---|---|---|---|---|---|---|
CD | Ia | 17 | Mean 13.9 ± 2.1 | 17 | Healthy controls | Lower bone mineral content/bone width | N/A | [96] |
CD | I | 123 | (Mean ± SD) Crohn 11.8 ± 2.9 (M), 11.9 ± 2.4 (F), UC 10.1 ± 2.8 (M), 11.7 ± 2.6 (F) | Crohn 43/39, UC 22/19 | Hologic database | Crohn, spine Z-score −1.44 ± 0.97 (M), −1.37 ± 1.22 (F)c. UC, spine Z-score −0.93 ± 1.10 (M), Z-score −0.56 ± 0.89 (F) | Male | [89] |
UC | Pb | Relatively young at diagnosis | ||||||
Healthy controls | ||||||||
No immunomodulator | ||||||||
CD | I | 18 | Mean (range) 12.9 (5.5–16.8) | 11/7 | GE Lunar Database | Body −0.24 ± 0.79 | Low BMI, weight, height, lean body mass | [90] |
Spine −0.99 ± 1.1** | ||||||||
Delayed puberty | ||||||||
Decreased physical activity | ||||||||
CD | I | 23 | Mean ± SD 12.58 ± 2.37 | 15/8 | Healthy controls Van der Sluis [80] | Body 0.36 ± 1.99 | Low BMI | [3] |
Spine −0.14 ± 1.04 | ||||||||
CD | I | 58 CD | GE Lunar Database | Body | Low BMI | [77] | ||
UC | 18 UC | CD −0.78 ± 1.02* | High IL-6 | |||||
49 Control | UC −0.46 ± 1.14 | |||||||
Control −0.17 ± 0.95 | ||||||||
CD | P | 47 CD | 46/27 | GE Lunar Database | dCD −2.0 (−3.8, −0.3) UC 1.2 (−3.2, 0.8) | Short stature | [97] | |
UC | 26 UC | Low BMI | ||||||
CD | P | 29 | Median (range) 15.18 (13.85–17.18) | 20/9 | GE Lunar Database | eBody −1.20 | Corticosteroids | [91] |
Spine −2.10 | ||||||||
CD | P | 28 CD | Median (range) 13 (5–18) | N/A | Faulkner [92] | 18/28 CD | N/A | [93] |
UC | 10 UC | 8/10 UC had “low Z-scores” | ||||||
CD | P | 119 | 72/47 | 38/119 < −2.0 | Weight and height Z scores, male sex | [94] | ||
81/119 < −1.0 | ||||||||
CD | I | 78 | 12 (5–18) | 44/34 | CHOP | pQCT of the tibia | Serum albumin, HCT | [71] |
Decreased trabecular BMD | ||||||||
Increased cortical BMD | ||||||||
Increased endocortical circumference | ||||||||
Decreased periosteal circumference | ||||||||
CD | P | 22 CD | 4–18 | 34/21 | Body SDS −0.95 | Corticosteroids, BMI SDS, Crohn disease | [95] | |
UC | 33 UC | Spine SDS −0.75 |
I incident cohort; P prevalent cohort; CD Crohns disease; UC ulcerative colitis
*P < 0.01
**P = 0.002
N/A not available
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
