Immunotherapy of Gynecologic Malignancies
PAUL J. SABBATINI  KUNLE ODUNSI
KUNLE ODUNSI  JACOBUS PFISTERER
JACOBUS PFISTERER  GEORGE COUKOS
GEORGE COUKOS
The natural clinical history of ovarian cancer makes it particularly suited for the evaluation of immune-based strategies. Although the majority of patients are diagnosed with advanced disease, 70% are in a complete clinical remission following initial cytoreductive surgery and platinum and taxane-based primary chemotherapy (1). However, only 30% of optimally debulked stage III patients will remain disease-free with a median progression-free interval of approximately 24 months (2). Despite the high relapse rate, many patients return to a complete or partial clinical remission following additional chemotherapy, but ever-shortening intervals of response are seen (3). Despite this chronic course of relapse and response, the median survival of optimally debulked patients exceeded 60 months in a study evaluating intraperitoneal (IP) therapy as part of primary treatment (4). Neither higher doses nor protracted schedules, nor non-cross-resistant consolidation chemotherapy has provided additional benefit. Ovarian cancer patients with minimal disease burdens are therefore appropriate candidates for the evaluation of immune-based strategies, which typically have excellent side-effect profiles.
With regard to other gynecologic malignancies, the majority of immune strategies being evaluated in patients with cervical cancer target human papillomavirus, and this subject is covered separately in the appropriate chapter (5). The evaluation of immune-based approaches for the treatment of endometrial cancer is limited but interest is growing (6). Targets such as human trophoblast cell surface marker (TROP-2), Wilms Tumor Gene (WT-1), and a variety of cancer testis antigens have been shown to be expressed in endometrial cancers which has prompted early stage trials with dendritic cell vaccines and adoptive cellular therapy as two examples (7, 8). The bulk of the clinical information available to date with regards to immunotherapy when one excludes cervical carcinoma, however, remains in patients with ovarian cancer.
CANCER IMMUNOLOGY: OVERVIEW
The immune system evolved to fight foreign invaders such as bacteria, viruses, and parasites. However, strong evidence suggests that it also plays a crucial role in controlling or rejecting incipient cancers. William B. Colley first observed regression in patients with sarcoma contracting “accidental erysipelas” as early as 1893 (9). Since patients with competent immune systems still develop cancers, and spontaneous remission of tumors is rare, enthusiasm for the effectiveness of immune system control has waxed and waned over the decades. The fact that animal models with a variety of deficiencies in immunologic components consistently develop carcinoma has allowed the concept of immunosurveillance to be sustained and developed (10, 11). Observations in patients with ovarian cancer strongly support a role for immune system involvement in patient outcome. For example, the 5-year overall survival in epithelial ovarian cancer has been correlated to the presence or absence of tumor infiltrating lymphocytes (TIL) (38% vs. 4.5%, p < 0.001) (12). A second study also showed improved survival in patients with increased frequencies of intraepithelial CD8+ TIL (55 months vs. 26 months; HR = 0.33; 95% CI, 0.18–0.60; p = 0.0003) (13). In contrast, patients with increased numbers of immune suppressive CD4+CD25HI regulatory T-cells (Tregs) have reduced survival (14). The process is made more complex when we recognize that not only can the immune system protect the host against tumor development, but it is thought to select cancer cells of lower immunogenicity which can escape early immunity due to changes in gene expression. This actually leads to an outgrowth of tumors with the capacity to escape recognition and this process has been termed immunoediting (15). The immunoediting or “immune sculpting” process, therefore, is responsible for shaping the immunogenicity of the tumors that will eventually form. Considering the effectors of the immune system in the context of both of these processes is important in order to develop immune directed therapies with the greatest chance of success (16, 17).
The mechanism by which tumors escape from immune surveillance is complex. An alteration in the function of almost every process discussed herein to facilitate immune activation has been postulated to participate in the ways tumors can evade immune detection. In some cases, antigen presentation is down regulated, or gene deletions or rearrangements may cause reduced expression of the MHC-I complex thus preventing T-lymphocyte activation (18, 19). Tumors can also secrete proteins that inhibit T-cell effector action or that promote the development of regulatory T-cells that suppress immune function (20). A recent observation showed that certain melanomas can actually remodel their stromal microenvironment so it resembles lymphoid tissue which recruits regulatory cells to promote tolerance and allow tumor progression (21). Other mechanisms include the downregulation of intracellular adhesion molecules (22), changes in molecules responsible for apoptosis signaling (23), or the development of peripheral tolerance (24). Based on the increasing number of interacting mechanisms with putative activity that allow immune escape, approaches directed against multiple mechanisms will likely be needed in order to eradicate immune tolerant tumor cells (25).
Innate Immunity
The immune system is broadly divided into 2 arms, innate and active immunity, but there are multiple examples of cross communication between them. Recent data, for example, show that neutrophil survival is influenced by T cell responses (26). Innate immunity is present at birth and does not require adaptation to react against microorganisms or tumors. It includes physical barriers (mucous membranes, skin), chemical components (compliment, hydrolytic enzymes), and multiple other cellular components. For example, natural killer cells are lymphocytes that are programmed to recognize and destroy tissues that have been altered or stressed, e.g., by viruses or by malignant transformation (27). They do not have antigen specific receptors but instead have inhibiting receptors that can recognize MHC class I molecules on normal cells, which prevents their activation (28). MHC I expression is aberrant or absent on many virus and tumor infected cells (29). Another component includes macrophages, which play many roles in immunity and inflammation, including production of a multitude of soluble secreted proin-flammatory proteins, which are growth factors for other cells of the immune system, for neovasculature, and for cancer cells. These growth factors include cytokines and chemokines, and they support the growth, movement, and survival of immune and inflammatory cells. Macrophages play important roles in tissue remodeling during wound healing, mediating inflammatory responses, sampling molecules from the environment through internalization, and presenting antigens to stimulate T cells. Macrophages can also play a counterproductive role through production of molecules that promote tumor growth and angiogenesis (e.g., vascular endothelial growth factor [VEGF] and basic fibroblast growth factor [FGF]) (30). Macrophages can also inhibit immune responses through production of cytokines and other molecules that down-regulate immunity, such as prostaglandins E2, arginase I, and transforming growth factor-β. Thus, the role of macrophages in tumors is complex: they provide an additional means of communication between traditionally defined innate and acquired immunity effectors (31). Dendritic cells have some properties that are similar to macrophages, including production of cytokines and sampling of molecules from the environment. Most importantly, dendritic cells are one of the key links between the innate immune system and the adaptive or acquired immune system through presentation of antigens (professional antigen-presenting cells) to initiate T-cell activation.
Adaptive Immunity
Adaptive immunity is characterized by adaptation to antigens, for example, antigens of infectious pathogens (or potentially by cancer). This arm of the immune system is not mature or activated at birth, but rather adapts through maturation in response to antigens of pathogens. It is characterized by receptors encoded by the immunoglobulin (Ig) gene family. These receptors have the capacity to rearrange, creating enormous diversity (>10 different receptors) (11–12). This provides a system to generate enormous specificity that recognizes and responds to a wide range of antigens that have not previously been encountered.
The two major cell types of acquired immunity are T lymphocytes (T cells) and B lymphocytes (B cells). T cells have the capacity to recognize antigens sequestered in different compartments within cells, for instance, antigens generated in the cytoplasm or nucleus by viral infections (32). T cells recognize antigens as short peptides, 8 to 16 amino acids in length. These peptides must be complexed with and presented by specialized antigen-presenting molecules, the major histocompatibility complex (MHC) molecules, to T-cell receptors. In humans, MHC molecules are the human leukocyte antigens (HLAs) expressed on virtually every cell in the body. On the other hand, B cells produce secreted antibodies that can recognize soluble and cell surface molecules. Both T cells and B cells initially develop with a limited range of receptors for immune recognition. However, in response to antigen (e.g., from a virus or cancer cell) presented by a professional antigen-presenting cell, T cells or B cells that have receptors with the best “fit” to the antigen itself (for B cells) or antigen/MHC complex (for T cells) are stimulated to proliferate and this subpopulation is quickly expanded (33).
Humoral Immunity
B cells recognize antigens usually in their natural configuration (32). An individual host has a repertoire of B cells that are capable of generating antibodies against the full range of pathogens encountered in the environment. To do this, the total population of B lymphocytes expresses a diverse repertoire of immunoglobulins. Each B cell expresses immunoglobulin against a single antigenic determinant, with the immunoglobulin expressed at the cell surface of the B cell where it acts as a specific receptor to transduce signals in response to that antigen (34). Once activated, the B cell is to create an individual monoclonal antibody (mAb). The diversity of specificities in different B cells is generated by rearrangements of the immunoglobulin genes, and new antibody specificities continue to be generated in response to new antigens (35). During development, B cells with high avidity immunoglobulins against ubiquitous self-antigens are eliminated. This elimination of B cells reactive with autoantigens is not absolute, however, as a broad array of antibodies against autoantigens is found in the blood of humans. Peripheral blood B cells consist of naïve and relatively short-lived B cells, long-lived memory B cells resulting from maturation in response to antigenic stimulation, and a small population of B cells expressing germ-line immunoglobulins that have not undergone rearrangement (which are found in the CD5+ B-cell population) (13).
B cells are mobile and after development in the bone marrow they migrate through the peripheral blood to B-cell-rich areas in lymphoid organs, e.g., the follicles of lymph nodes, spleen, and gastrointestinal tract. Many B cells continue recirculating in the blood. If cognate antigen is encountered in lymphoid organs, the B lymphocyte migrates to the T-cell-rich areas where appropriate T-cell help can be provided to promote increased antibody diversity and increased affinity through immunoglobulin gene rearrangements (36). This T-cell help does not have to be induced by the same antigen. Chemical conjugation of the antibody-recognized antigen to highly immunogenic bacterial or xenogeneic proteins or, alternatively, expression of the antigen in bacterial or viral vectors, is widely used approaches to ensure adequate T-cell help in vaccines. The result of T-cell help is generation of plasma cells, the most mature form of B cell with the highest capacity for antibody production. In addition, T-cell help promotes formation of germinal centers in lymphoid organs where hypermutation in immunoglobulin genes and class and subclass switching occur to generate antibodies with higher affinities for antigen. Class switching refers to changes in antibody class during an immune response, with the IgM class of antibodies appearing first, generally followed by antibodies of the IgG class (different subclasses of IgG antibodies have different blood half-lives and different capacities for effector functions, such as fixation of complement or binding to Fc receptors). The consequence is plasma cells secreting increasingly higher affinity IgG antibodies as the immune response matures over time (37). In addition, some B cells that generally recognize nonprotein antigens, such as carbohydrate or glycolipid antigens, can be stimulated to proliferate in the absence of T-cell help. The immunoglobulin variable region (called the Fv region) determines antibody specificity and is located in the Fab domain of immunoglobulins. This region mediates effective binding to antigens. However, the constant region (Fc), where antibody class and subclass are determined, is equally critical. Binding of antibody to antigen results in a conformational change in the Fc portion, leading to activation of several effector mechanisms, including complement activation (discussed below). IgM antibodies are synthesized early in the response. If T-cell help is available, antibody responses mature through immunoglobulin gene rearrangements into the higher affinity IgG classes. These are capable of improved binding to antigen as well as receptors on the bone marrow-derived cells for the Fc domain, expanding potential effector functions. The responses to most nonprotein antigens are IgM class and generally do not mature to IgG responses. The IgM pentamer structure is specialized to increase avidity of binding to multimeric antigens and to efficiently activate effector functions such as complement (38). Activation of complement, which includes blood components with different enzymatic properties, results in opsonization (coating of pathogens by complement components), recognition by complement receptors on macrophages, monocytes, neutrophils, and dendritic cells, with subsequent activation of these cells leading to phagocytosis and/or killing. In addition, complement can form a membrane attack complex, which creates holes in membranes of target pathogens and cancer cells, producing complement-dependent cytotoxicity (CDC) (39,40). IgG antibodies are synthesized following immunoglobulin gene rearrangements, with switches in Fc domains, as the B cell matures in response to T-cell help. IgG antibodies usually have higher affinity, and can be found in the extracellular space as well as in the blood. IgG1 and IgG3 antibodies in humans are especially effective at activating complement and also at sensitization of pathogens for killing by NK cells, macrophages, and other cells with complement receptors and immunoglobulin Fc receptors (41).
Opsonization for ingestion and destruction by phagocytes can occur through complement activation, but also occurs directly as a consequence of engagement of Fc receptors on phagocytic cells. Antibodies complexed to antigen bind to Fc receptors, which can lead to activation signals through activating Fc receptors (e.g., FcRIII), but activation can be countered by IgG binding to the inhibitory Fc receptor, FcRIIB. Fc receptors, which are bound effectively by IgG1 and IgG3 subclasses of human antibodies, are expressed on monocytes, macrophages, NK cells, neutrophils, mast cells, and other cells. Cross-linking of Fc receptors leads to activation of the cells, in some cases leading to antibody-dependent cell-mediated cytotoxicity (ADCC) of tumor cells through production of cytotoxic molecules, such as perforin and granzyme by NK cells and reactive molecular species by macrophages (42–44). Monoclonal antibodies are commonly used for cancer therapy. Antitumor effects can be mediated in part by antibody binding to critical molecules on the surface of tumor cells, for example by inhibiting tumor cell attachment or growth receptors. However, generally interactions of antibody with cell antigen are not very effective unless Fc receptor–mediated effector mechanisms are also activated.
Cellular Immunity
Tlymphocytes recognize processed (digested) molecules that complex with MHC molecules within antigen-presenting cells. The antigen/MHC molecules are then trafficked to the cell surface for recognition by T-cell receptors, which are encoded by genes of the immunoglobulin family. Similar to generation of antibodies, great diversity of T-cell receptors is generated by rearrangements of these immunoglobulin family genes. Each monoclonal T-cell receptor must bind to its cognate antigen/MHC complex (CD4 cells bind to MHC class II molecules, and CD8 cells bind to MHC class I molecules) presented on the surface of antigen-presenting cells (45,46). Signaling from the T-cell receptor following engagement of antigen/MHC complex is insufficient to activate the T cell. Additional signals are required (co-stimulatory signals or “signal 2”). The most important co-stimulatory signal in T cells comes from the T-cell surface molecule CD28, which engages B7 molecules (CD80 and CD86) on antigen-presenting cells (47–49). Engagement of T-cell receptor by antigen/MHC in conjunction with CD28 engagement of B7 is sufficient to activate naïve T cells. Within several days following T-cell activation, a second molecule, CTLA-4, appears on the T-cell surface to provide a brake to the T-cell response. CTLA-4 also binds B7 molecules, but with much higher affinity, therefore displacing CD28 activation signals. CTLA-4 signaling leads to down-regulation of the T-cell response. The manipulation of CTLA-4 activity has recently been shown to have therapeutic efficacy with recent data showing tumor responses in patients with melanoma (50,51). A variety of other co-stimulatory molecules are up-regulated on the surface of activated T cells, including OX40 and 4-1BB, which promote survival of T cells and help generate long-lived T-cell memory responses (52,53). Once T cells are activated by professional antigen-presenting cells (primarily dendritic cells), they gain a variety of effector functions, including the production of cytokines and cytotoxic molecules, which lead to death of target cells.
Antigen-Presenting Cells
Dendritic cells are the prototype professional antigen-presenting cells and are the critical link between innate immunity and acquired immunity. These are phagocytic cells that sit on epidermal surfaces, including skin and mucosal membranes, constantly sampling their environment to search for infectious organisms. Although dendritic cells continuously ingest molecules from their environment, uptake of antigen is insufficient to activate dendritic cells. Rather, dendritic cells have a set of receptors, most notably the toll-like receptors (TLRs), which can recognize lipid-containing molecules and CpG-rich DNA and poly-U RNA sequences produced specifically by microbial organisms. Engagement of TLR signals for activation of the dendritic cell, with increased expression of MHC and B7 molecules, and movement of cells with captured antigen to draining lymph nodes. It is in draining lymph nodes that dendritic cells activate appropriate T cells that recognize that particular antigen presented by MHC molecules. Subsequently, activated T cells can then travel from the draining lymph node to distant sites of infection or tumor to carry out the effector functions. One of the central problems for cancer immunology is that dendritic cells may ingest and process cancer antigens, but without activation through TLRs or other receptors, the dendritic cells remain incapable of activating T cells (because of insufficient expression of co-stimulatory molecules, such as B7) and do not move to draining lymph nodes. In fact, insufficiently activated dendritic cells presenting antigens can induce anergy, a form of immune tolerance where T cells become paralyzed and incapable of responding to cognate antigens. This is one of the mechanisms used to maintain immune tolerance to self to prevent autoimmunity, but also presents a major hurdle for cancer immunity. Cancer cells do not have any readily apparent mechanism to activate dendritic cells, although several self molecules, including heat shock protein, hyaluronate, and uric acid crystals have been suggested to cause activation.
Helper and Regulatory T Cells
Several types of T cells are activated by dendritic cells, and which type is influenced by whether antigens are presented by MHC class I or MHC class II molecules on antigen-presenting cells. MHC class I molecules complexed with antigen stimulate CD8+ T cells that are cytotoxic and kill target cells (infected cells or tumor cells). Antigens presented by class II MHC molecules stimulate CD4+ helper T cells. Helper T cells produce chemokines and cytokines to help recruit and orchestrate other components of the immune system. Helper T cells come in several types and different cytokines in the milieu determine what type of T cell is generated. Each type of helper T cell mediates different types of immune responses with different characteristics. Th1 T cells produce interferon (IFN)-γ to activate cytotoxic T cells and macrophages for cellular immune responses. On the other hand, Th2 helper T cells produce interleukin (IL)-4 and other cytokines that favor antibody responses, or humoral immunity. The newly discovered Th17 CD4+ T cell produces IL-17 to mediate inflammatory autoimmune diseases, such as arthritis, colitis, and encephalitis, and may play a role in cancer pathogenesis, either by promoting cancer progression or by destroying tumors. Another type of CD4+ T cell restricted by MHC II presentation negatively regulates immune responses. These are regulatory T cells, or Tregs. Tregs recognize self antigens, are dependent on IL-2, and produce inhibitory molecules such as IL-10 and transforming growth factor-β. Th1 cellular immunity may be particularly effective at attacking tumors in tissues, while humoral immunity may have an advantage in controlling circulating metastatic tumor cells. Infiltration of ovarian cancers and other cancers by Treg cells is associated with a poorer prognosis (50,54).
CANCER IMMUNITY AND IMMUNOTHERAPY
Careful studies over the past 3 decades have shown that the immune system can recognize and destroy cancers, usually involving interactions between multiple arms of the immune system. For instance, simple recognition by T cells, antibodies, or NK cells is usually not sufficient to reject cancers. Cytotoxic T cells and NK cells produce soluble and cell surface molecules that induce death of target tumor cells. Helper T cells can produce cytokines and chemokines that not only recruit cytotoxic T cells or B cells to make antibodies, but also inflammatory cells which mediate tissue destruction. Antibodies can activate NK cells, macrophages, or other cells that have receptors for the antibodies’ Fc domain, leading to activation of the recruited cells, a mechanism implicated in the antitumor effects of monoclonal antibody therapies. Antibodies can also activate complement proteins in the blood that can directly kill tumor cells and activate inflammatory cells at the tumor site.
The abundant evidence that the immune system can recognize antigens on tumor cells and that immunity is sufficient to destroy tumors, provides the impetus to continue and develop strategies for immunotherapy. These approaches can be broadly divided into three groups. First, immune modulation uses more broad approaches to treat cancer. Traditional examples include cytokines such as IL-2, IL-12, or interferons, and more recently more targeted approaches such as those interfering with CTLA-4 or PD-1 (51). Secondly, passive therapy refers to the transfer of specific components from the acquired immune system to the host with cancer. The best examples are monoclonal antibodies directed against antigens expressed on the surface of cancer cells such as rituximab against the CD20 antigen, or trastuzumab (Herceptin) directed towards the HER 2 receptor in breast cancer. It should be noted that although the antitumor effects of trastuzumab in part probably involve inhibition of signaling by the HER2 tyrosine kinase oncogene, evidence strongly supports a major role for immune activation of Fc receptor–positive cells by both rituximab and trastuzumab (55). Another example of passive immunotherapy is adoptive cellular therapy, where cells from the blood or bone marrow donor are purified, cultured, and/or manipulated outside the body and reinfused into the same patient (autologous) or a different patient (allogeneic). Cellular therapy has particularly focused on T cells and more recently on NK cells. Finally, active immunization refers to vaccines that trigger the patient to make an immune response. Both passive and active immunotherapy must be directed against specific antigens on cancer cells, and the notion of an integrated immune response with effectors to include antibodies and T cells as well as some component of immunomodulation is increasingly becoming the goal of immunotherapeutic strategies (56).
Antigens for Immunotherapy |
Antigen Category | Representative Examples |
Differentiation antigens | |
Mutational antigens | |
Amplification antigens | |
Slice variant antigens | |
Glycolipid antigens | MUC1 (66) |
Viral antigens | HPV (67) |
Cancer testis antigens |
With regards to potential targets, ongoing advances utilizing newer technologies including serological analysis of recombinant cDNA expression libraries (SEREX) (57,58), robust applications of bioinformatics (59) and seromic profiling techniques (60) have allowed the further characterization of tumor associated antigens in multiple tumor types. There are over 2000 candidate tumor associated antigens and they are generally classified as follows: (61) (a) differentiation antigens (62), (b) mutational antigens (63) (that are altered forms of proteins), (c) amplification antigens (64), (d) splice variant antigens (65), (e) glycolipid antigens (66), (f) viral antigens (67), and (g) cancer-testis (CT) antigens (68). Representative examples of each group are seen in Table 15.1.
In addition to selecting the appropriate targets, it is necessary to select the strategy to be used for immunotherapy. Multiple approaches have been considered from giving the effector cells directly to include antigens of a variety of types given alone or with adjuvants, modified or unmodified tumor cell lysates can be administered (autologous or allogeneic), dendritic cells can be primed with a variety of agents, tumor hybrids with antigen-presenting cells can be made, or DNA alone or in a recombinant fashion can be administered. A sample of cancer vaccine strategies are listed in Table 15.2.
Examples of Vaccine Approaches |
I. Antigens alone with or without adjuvants
Peptides
Protein
Gangliosides
Immunoglobulin idiotypes
II. Dendritic cells
Peptides, immunoglobulin idiotype
Tumor lysates
DNA or RNA
Protein
III. Tumor cells unmodified or modified
Autologous
Allogeneic
Mixed autologous-allogeneic
IV. Tumor-APCs hybrid
V. DNA alone (naked DNA), recombinant viruses (adenovirus, vaccinia, and others)
A critical component of any immune approach is a consideration of immune escape. Tumors escape destruction by the immune system via a variety of active, regulatory mechanisms. These include downregulation of MHC and tumor antigen loss (69), stimulation of the inhibitory receptors on T-cells such as CTLA-4 (70), PD-1 and LAG-3 (71), tumor overproduction of indoleamine 2,3-dioxygenase (IDO) (72), induction of increased tumor infiltration by regulatory CD4+CD25+ T cells (Tregs) (73) and also certain types of natural killer T (NKT) cells that inhibit tumor immune destruction (74). Overcoming these mechanisms is the goal of immunomodulation.
IMMUNOMODULATION
Multiple potential difficulties remain in developing new immunotherapy approaches, including insufficient activation of dendritic cells by cancer cells, inhibition of responses by Treg cells, and both intrinsic and extrinsic mechanisms that down-regulate T-cell responses, e.g., signaling through CTLA-4. These checkpoints all participate to dampen immune responses against cancer. Modulating immune check points by activation of effector cells, depletion of Tregs or activation of professional APCs could substantially improve the therapeutic efficacy of vaccines or adoptively transferred T cells. The development of functional antibodies is now enabling effective immunomodulation as shown in Figure 15.1. Passive therapy with monoclonal antibodies or with adoptive cellular treatments may also be able to bypass some of these checkpoints.
Dendritic Cell Activation
The main mechanism of immune stimulation by CD 40 ligands is activation of DCs resulting in increased survival, upregulation of costimulatory molecules, and secretion of critical cytokines to T cell priming such as IL-12. This promotes antigen presentation, priming and cross priming of CD4+ and CD8+ effector T cells (75). However, agonistic anti-CD40 antibody is best used in combination with vaccines or Toll-like receptor agonists (75,76), because alone it can accelerate the deletion of tumor-specific cytotoxic lymphocytes (77). Additional value of CD40 ligation is provided by the fact that ovarian cancers, like many tumors, express the CD40 receptor (78–81) and respond to CD40 agonists with apoptosis and growth inhibition in vitro and in vivo (80,82,83).
Effector T-cell Activation
T-cell activation is triggered through the T-cell receptor by recognition of the cognate antigen complexed with MHC. This activation is regulated by complex signals downstream of CD28 family immune receptors, which includes costimulatory (CD28 and ICOS) and inhibitory receptors (CTLA-4, PD-1 and BTLA). PD-1 and CTLA-4 are induced on T cells following a TCR signal, and result in cell cycle arrest and termination of T-cell activation. The use of blocking CTLA-4 or PD-1 mAbs can sustain the activation and proliferation of tumor-specific T cells, preventing anergy or exhaustion and thereby allowing the development of an effective tumor-specific immune response.
CTLA-4 Blockade
CTLA-4 is the best characterized among the inhibitory B7 receptors. As with CD28, CTLA-4 also binds CD80 and 86 expressed by APCs, but provides inhibitory signals to the T cell, serving as a negative feedback loop. In animal models, CTLA-4 proved critical in the normal functioning immune system. CTLA-4 knockout mice develop uncontrolled lymphoproliferation with early lethality. Furthermore, administration of CTLA-4 blockade significantly enhanced antitumor immunity in a variety of mouse models and with various therapeutic combinations including vaccine, TLR agonist, chemotherapy and radiation therapy. The mechanism by which CTLA-4 blockade induces tumor regression is not fully understood. Although Treg constitutively express CTLA-4, CTLA-4 blockade had no effect on the number or function of Treg in cancer patients. It seems that CTLA-4 blockade activates directly CD4 and CD8+ T effector cells by removing an inhibitory checkpoint on proliferation and function (84). The combination of direct enhancement of Teff cell function and inhibition of Treg activity is essential for mediating the full therapeutic effects of anti-CTLA-4 antibodies during cancer immunotherapy (85).
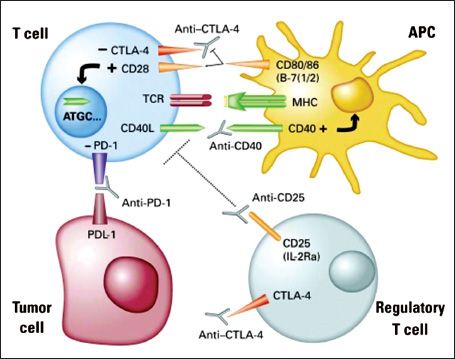
FIGURE 15.1. The modulation of immune checkpoints.
Source: From Kandalaft, LE, Powell DJ Jr, Singh N, et al. Immunotherapy for ovarian cancer: what’s next? JCO 2011;29:925–333, with permission.
Based on these data, several clinical studies have been undertaken to assess the efficacy of a CTLA-4 antagonist antibodies in the setting of human cancer. The majority of clinical data to date have emerged from studies in patients with melanoma (reviewed in (86)). Phase II studies with ipilimumab monotherapy showed objective responses at a dose of 3 to 9 mg/kg every 3 weeks, with 10 mg/kg showing the best risk-benefit profile. Ipilimumab (3 mg/kg every 4 weeks x 4) administered in combination with chemotherapy (dacarbazine) resulted in enhanced objective response rates without added toxicity. Ipilumimab has also been combined successfully with IL-2 (720,000 U/kg every 8 hours x 15 (OR rate of 22% – 3 CRs) or melanoma vaccine (OR rate of 13% – 2 complete responses). In addition, tremelimumab has also produced OR and CRs in melanoma patients. To date, CTLA-4 antibody has not shown superiority over standard chemotherapy trial in treatment-naive melanoma patients in a phase III randomized clinical, which was discontinued after interim analysis. In melanoma patients, the most common grade 3 to 4 adverse events were colitis/enterocolitis (increased frequency of diarrhea/stools), dermatitis, and hypophysitis.
Eleven patients with ovarian carcinoma, previously vaccinated with GM-CSF modified, irradiated autologous tumor cells (GVAX), received ipilumimab (1 month to 3 years following GVAX). Significant antitumor effects were observed in a minority of the patients. One patient achieved a dramatic fall in CA-125 levels several months after an initial dose of ipilumimab; although the response was not maintained, a second infusion resulted in a more rapid decline in CA-125 values. Nine additional infusions of the anti-CTLA-4 antibody spaced at 3- to 5-month intervals over nearly 4 years have maintained profound disease control, with grade 1 rash as the only adverse event. An objective radiographic response was noted in parallel with the reduction in CA-125. Moreover, this patient’s course was initially characterized by a remarkable periodicity to the rise and fall of CA-125 levels, but this gradually dampened as a function of time and/or prolonged therapy, demonstrating a lack of acquired resistance to ipilimumab commonly observed with cytotoxic chemotherapy (87). In contrast to the melanoma cohort, a single dose of 3 mg/kg Ipilumimab triggered 2 cases of grade 3 gastrointestinal inflammation among the 9 ovarian carcinoma patients, consistent of significant diarrhea. Endoscopic biopsies revealed mucosal damage associated with abundant granulocytes, macrophages, CD4+ and CD8+ T cells, and FoxP3+ Treg. The remaining seven patients showed only minor inflammatory toxicities including papular rash, or urticarial-like reactions at sites of prior vaccination (87). Tumor regression in these patients correlated with the CD8+/Treg ratio, suggesting that other forms of therapy that target Treg depletion may provide a highly effective form of treatment when combined with the tumor vaccine and CTLA-4 antibody arsenal (87).
PD-1 Blockade
Programmed death receptor-1 is a negative regulator of cell activation, primarily expressed on both B and T lymphocytes, which binds PD-L1 and PD-L2 ligands. PD-L2 is restricted to professional antigen-presenting cells, while PD-L1 is expressed on many tissues, and plays a pivotal role in maintaining peripheral tolerance. Importantly, cancer cells have adopted expression of PD-L1 into their immunosuppressive arsenal (88) and a variety of epithelial cancers express PD-L1 (89–91). It has also recently been shown that ovarian carcinoma cells as well as tumor-infiltrating tolerogenic DCs and myeloid derived suppressor cells express PD-L1 (92,93). Furthermore, expression levels correlate with disease course. Although PD-1 signaling can occur at very low levels of receptor expression, elevated levels of PD-1 have recently been attributed functional significance. During conditions of chronic antigen persistence (such as in chronic viral infections), antigen specific effector T cells expressing high levels of PD-1 were demonstrated to be functionally exhausted, i.e., unable to proliferate, secrete IL-2, or kill target cells (94). It has been shown recently that tumor infiltrating lymphocytes in metastatic melanoma and other tumors expresses higher levels of PD-1 than CD4+ and CD8+ cells in the peripheral blood of healthy patients and exhibit phenotypic and functional characteristics of exhausted T cells with impaired effector function. These findings suggest that the tumor microenvironment can lead to up-regulation of PD-1 on tumor-reactive T cells and contribute to impaired antitumor immune responses (95). Indeed, constitutive or inducible expression of PD-L1 by tumors conferred resistance to immunotherapy in mice related to failure of antigen-specific CD8+ CTLs to destroy tumor cells but without impairment of CTL function (96).
Following evidence that PD-1 blockade through gene therapy approaches in the tumor microenvironment activates antitumor immunity (97), antibodies blocking PD-L1 or PD-1 were found to profoundly enhance the efficacy of immune therapy (96,98). Anti-PD-1 antibody has been combined with cell-based vaccines and demonstrated increased infiltration, potency and duration of activity of CD8 T cells in the tumor microenvironment in the setting of B16 melanoma and CT26 colon carcinoma in mice.
Despite their similarities, the regulatory roles of CTLA-4 and PD-1 may differ substantially in cancer patients. First, Even though PD-1 and CTLA-4 ligation efficiently block T-cell responses, the homeostatic function of the 2 inhibitory receptors differs. For example, CTLA-4 ligation does not inhibit PI3-kinase signaling, while PD-1 engagement blocks the induction of PI3K activity. PD-1-mediated signaling blocks T-cell activation more effectively than CTLA-4-mediated signaling. In addition, PD-1 signaling may be related to T-cell apoptosis. Second, whereas the ligands for CTLA-4, CD80 and CD86, are primarily expressed by mature antigen-presenting cells, PD-L1 (on tumor cells or myeloid cells) and PD-1 (on effector T cells) are significantly upregulated in the tumor microenvironment. Thus, the effects of PD-1 blockade may be more pronounced in the tumor than in periphery. A phase I study using PD-1 blocking antibody showed the antibody to be safe and well tolerated in patients with hematologic malignancies. No single maximum tolerated dose was defined in this study. Clinical benefit was observed in 33% of the patients with one complete remission (99).
T Regulatory Cell Depletion
CD4+ CD25+ Foxp3+ T regulatory cells are responsible for maintaining peripheral tolerance by inhibiting T-cell activity. A number of Treg-depleting strategies have been investigated (100–104). An example is the use of low dose oral or intravenous cyclophosphamide (105,106). Other strategies for Treg depletion are through targeting the IL-2 receptor alpha chain, also known as CD25. In mouse models, the use of anti-CD25 monoclonal antibody before vaccination led to complete tumor rejection and establishment of long-lasting tumor immunity with no autoimmune complications (107,108). Administration of anti-CD25 antibody linked to a potent pro-inflammatory toxin showed significant but transient reduction in CD4+ CD25+ Treg cells in patients with metastatic melanoma (109). Another clinical approach of targeting CD25 is through Denileukin diftitox, a fusion protein of IL-2 and diphtheria toxin that targets CD25-expressing cells used in patients with melanoma, ovarian cancer and renal-cell carcinoma (110–113). Although effective in short-term infusions, these conjugates are quite immunogenic and induce neutralizing antibodies, which hamper their long-term application.
Another agent is Daclizumab, which is an FDA-approved humanized IgG1-kappa mAb that binds specifically to CD25 (114). It has been used in autoimmune disorders (115,116), acute graft-versus-host disease (117), and in cancer patients with CD25+ T-cell malignancies (118). The advantage of Daclizumab is that it is well tolerated, and has a half-life of 20 days (119). In a recent study, Daclizumab was used in a single dose of 1 mg/m2 prior to hTERT peptide vaccine for metastatic breast cancer. Total CD4+CD25+ and CD4+CD25+FoxP3+ cells remained suppressed for several weeks after a single infusion.
Cytokines
Cytokines play important roles in immune modulation. Many cytokines, including IL-2, IL-3, IL-4, IL-6, IL-10, IL-12, tumor necrosis factor-α (TNF-α), macrophage colony-stimulating final factor, and IFNs, have been studied for their roles in tumor treatment. Some cytokines (IL-2 and IFN-α) have been approved by the U.S. Food and Drug Administration (FDA) specifically for the treatment of melanoma and renal-cell carcinoma though enthusiasm for use has waned given the systemic toxicity profile (120,121). Ovarian cancer ascites is rich with cytokine expression and the study of the ascitic fluid should help to better understand the immune interactions between tumor and host which is complex. A recent analysis showed ascites to be high in pro-inflammatory cytokines IL-6, IL-8 and the immune suppressive cytokines IL-10, CCL22 and TGF-B in most samples. Interestingly, high IL-6 expression predicted residual disease after debulking and was highest at recurrence (122).
Interferons
Although the full extent of action of IFN in patients with ovarian cancer is unknown, several mechanisms have been proposed: (a) stimulation of NK cells and macrophages, both of which are known to have antitumor properties (21); (b) antiangiogenic effects; and (c) inhibition of expression of dysregulated oncogenes (such as HER2/neu) and thereby improving the responsiveness of cisplatin-resistant cells (123). A Gynecologic Oncology Group Study evaluated administration of systemic IFN-α to patients with advanced ovarian cancer where patients with measurable disease showed a low response rate of approximately 10% (124). The role of intravenous IFN-α as a maintenance treatment after initial surgical resection and chemotherapy was also evaluated by a randomized phase III study and showed no benefit (125). Systemic IFN-α was also associated with frequent dose-limiting toxicity. Because of the poor response rate and frequent toxicity of systemic administration, further studies were focused on IP administration of IFN-α. Multiple clinical trials with IP IFN therapy were carried out leading to a randomized trial of adjuvant intraperitoneal α-interferon in stage III ovarian cancer patients who had no evidence of disease after primary surgery and chemotherapy by the Southwest Oncology Group (126). The trial was closed early but there was no difference between the study arms with regard to progression-free survival (p = 0.56). In cervical cancer, monotherapy with IFN-α had minimal activity (127). Studies evaluating intralesional interferon in HPV associated diseases (cervix and vulvar cancer) have waned as directly targeted immunologic approaches against the virus have emerged. Nonetheless, a recent study utilizing intralesional IFN = α 2 b in high grade cervical intraepithelial neoplasia showed a 60% response rate. Treated patients also expressed more Th1 (IFN-γ, TNF-α, IL-2) cytokines, with a significant reduction in the viral load of high-risk HPV (p = 0.0313) (128).
Interleukin-2
IL-2 is a T-cell growth factor that plays a central role in the immune system with data showing IL-2–induced CD4+ T cell expansion in patients with HIV infection (129) as well as clinical responses in renal cell cancer and malignant melanoma resulting in an indication for treatment by the U.S. Food and Drug Administration for these tumors (120,121). It has been shown to activate tumor-associated macrophages and upregulate IFN-stimulated genes (130). Based on the toxicity of intravenous IL-2 administration, studies in ovarian cancer focused on IP administration. A phase I-II study (131) of IP IL-2 in patients with laparotomy-confirmed persistent or recurrent ovarian cancer after ≥6 courses of prior platinum-based chemotherapy had shown an overall response rate of 25.7%, with an overall 5-year survival probability of 13.9%. A more recent study shows that IP IL-2 administration in patients with ovarian cancer increases CD4+CD25 high T-cells which highly express FOXP3 and suppress Treg cells. This suggests that IL-2 has a critical role in maintaining the Treg pool as well as enforcing Treg cell tumor trafficking in humans and may lend itself to combination studies of other agents targeting different immune checkpoints (132,133). A disadvantage of frequent administration of intravenous high-dose IL-2 is the occurrence of dose-limiting side effects. Therefore, delivery of IL-2 from an expression plasmid has been evaluated for the treatment of ovarian cancer. IP treatment of ovarian tumors with an IL-2–expressing plasmid resulted in an increase in local IL-2 levels, a change in the cytokine profile of the tumor ascites, and a significant antitumor effect in a mouse model (134). A recent in vitro study in colorectal cancer showed the ability of plamids expressing murine IL-2 to enhance the antitumor activity of viral vector immunization against a cancer mucosa antigen showing the tolerability and possibility of combining plasmid-derived IL-2 production with other immunotherapeutic strategies as well (135).
Interleukin-12
IL-12 is a cytokine mainly produced by activated monocytes, tissue macrophages, and B cells. It can induce IFN-γ and together with IL-2 becomes a potent activator of cytotoxic T lymphocytes and NK cells (136,137). Whereas IL-4 and IL-10 mediate the development of Th2-type immunity, IL-12 initiates the differentiation to the Th1 phenotype. In addition, IL-12 production can be negatively or positively regulated by cytokines. For example, IL-10 and IL-4 have been shown to inhibit the production of IL-12 (138), whereas IL-2 and IFN-α enhance its production. In addition, IL-12 has potent antimetastatic and antitumor effects in several murine tumor models, as well as in human tumor cells in vitro and in vivo (139). Ascitic IL-12 had been shown to be an independent prognostic factor for adverse outcome in ovarian cancer (140) though later there was uncertainty over this finding as a result of a possible statistical flaw (141). A phase II GOG clinical trial (142) evaluated intravenously administered recombinant human IL-12 in patients with recurrent or refractory ovarian cancer. Myelotoxicity and capillary leak syndrome were the major adverse events. Partial response rate was on 3.8% with no complete responders. Recent focus has shifted to evaluating the administration of IL-12 expressing DNA. Two studies have evaluated this strategy in patients with malignant melanoma with patients showing response of greater than 30% in 5 of 12 patients receiving intratumoral injections; or CR in 2 of 19 and PR or SD in 8 of 19 patients using electroporation of plasmid coding IL-12. More investigation in this area including other tumor types is warranted and ongoing (143,144).
ANTIBODY MEDIATED THERAPY OF CANCER
Antibodies are the primary mechanism for eliminating infectious pathogens from the bloodstream. The effect of most commonly used vaccines against infectious agents is thought to be primarily a consequence of antibody induction with subsequent complement mediated inflammation and cytotoxicity (145). In addition, tumor antigen-specific autoantibodies are found in the sera of patients with solid tumors and generally increase with tumor burden (146,147). As one example, the presence of p53 autoantibodies was detected in 41.7% of patients with serous cancer, and detectable p53 antibodies were associated with improved overall survival (p = 0.04; HR = 0.57; 95% CI, 0.33, 0.97) (148). It is unknown whether this finding represents a biomarker for prognosis, or if the antibodies exert immunologic control of tumor. Another example is found in the presence of natural antibodies (i.e., prior to vaccination or administration) in paraneoplastic syndromes. In a series of 100 patients with small cell lung cancer, patients with autoantibodies causing the Lambert-Eaton myasthenic syndrome had improved survival (19.6 months) versus those who were antibody negative (8.9) (149). In platinum resistant ovarian cancer patients, another study showed that anti-MUC1 IgM antibodies correlated with overall survival at both early (p = 0.052) and late (p = 0.009) time points (150). These results provide the possibility that functionally effective antibodies may confer a survival advantage. The possibility that these represent a biomarker for better outcome instead cannot be excluded from this data. The ability of antibodies to mediate protection from tumor recurrence has also been suggested in experimental animal models (151). The administration of monoclonal antibody 3F8 against GD2 or induction of GD2 antibodies by vaccination after challenge with EL4 lymphoma (which expresses GD2) showed prolonged survival in mice receiving antibody (p < 0.004) or vaccination (p < 0.008) (151). Trials in gynecologic cancers have included the administration of murine or humanized antibodies directly targeting tumor associated antigens, indirect approaches utilizing anti-idiotypic antibodies, or antibodies conjugated to radionuclides, cytokines, or other immunotoxins. The greatest success in the antibody mediated treatment of cancer to date has come from a series of monoclonal antibodies against antigens expressed at the cell surface of cancer cells with clinical efficacy in both the advanced disease and adjuvant settings. Monoclonal antibodies are produced by hybridoma cells. They has evolved from simple murine derived antibodies with recombinant engineering techniques to now permit monoclonal antibodies of various sizes, configuration, valence, and target effector functions (152). Examples with proven efficacy continue to grow to include an ever increasing list such as rituximab, ibritumomab, and tositumomab for B-cell lymphomas; gemtuzumab for acute myelocytic leukemias; alemtuzumab for chronic lymphocytic leukemias; cetuximab for colorectal and head and neck squamous cell carcinomas; and trastuzumab for breast cancer (152,153). A variety of mechanisms of action have been postulated: (a) monoclonal antibodies can mediate effector cells by having an activating or suppressive function in pathways such as with CTLA-4 (51), (b) they may target tumor supportive molecules such as VEGF with bevacizumab (154), or (c) they may recognize tumor specific receptors and by binding may activate or repress signaling pathways. Preclinical studies suggest that antibodies may have the most efficacy in the adjuvant setting. Since many gynecologic cancer patients are initially rendered free of detectable disease by surgery and/or chemotherapy after initial diagnosis, the administration of effective mAbs or vaccines with antibodies as part of the effector repertoire would have broad applicability. There are a variety of potential antigen targets for antibodies on gynecologic cancers. Screening for this purpose was traditionally limited to surface antigens since antibodies recognize antigens primarily at the cell surface. A previous study screened a series of 40 mAbs against 25 antigens common antigens that are potential targets for antibody mediated immunotherapy as listed in Table 15.3 (66,155,156). The antigens expressed by ovarian and endometrial cancers are similar, and quite different from those expressed by melanomas, and similar to, although not the same as, those expressed by prostatic cancers. The 18 excluded antigens (including CEA and HER2/neu) were expressed in 1 or 0 of 5 specimens. The expression of these antigens on normal tissues is essentially restricted to apical epithelial cells at luminal borders, a site that appears not to be accessible to the immune system. In addition to these overexpressed tumor associated antigens, other targets have included over-expressed growth-factor receptors (e.g., HER2/neu [erbB2], epidermal growth-factor receptor [EGFR], vascular endothelial growth factor [VEGF]); and mutated tumor-suppressor genes (e.g., p53 and BRCA-1). Binding of mAb to these antigens may have antitumor effect generated through a variety of possible mechanisms including: (a) antibody-mediated recruitment of human effector mechanisms in situ, ADCC against the tumor cells; (b) development of tumor-specific cytotoxic T lymphocytes (CTLs); (c) activation of the complement system; (d) stimulation of granulocytes cytotoxic to the cancer cells; and (e) induction of an anti-idiotypic antibody that can elicit active immunity.
Potential Antigens for Antibody Directed Therapy |
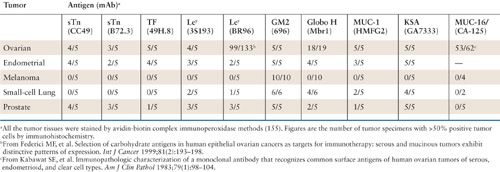
The extensive expression of CA-125 in over 80% of serous and endometrioid ovarian cancers has been well documented since the early 1980s (157). Following the successful cloning of CA-125 and identifying it as a complex mucin, it has been termed MUC-16 (158,159). Multiple efforts to target MUC-16 as a logical target for immunotherapy are underway.
Antibody Conjugates
In an attempt to add to the efficacy of unconjugated antibodies, efforts have been made to optimize the antitumor activity by conjugating with radionuclides, toxins, cytotoxic drugs, or second antibodies. The choice of conjugates and specific antibodies is influenced by features such as antigen internalization, lysosomal degradation, shedding, and heterogeneity of expression. The mechanism can be complex in that for some internalization is a prerequisite for cellular toxicity; while in others it may result in reduced efficacy and increased host toxicity due to intracellular catabolism (160).
Radionuclides
A variety of radionuclides have been conjugated with antibodies for imaging and treatment. The optimal radionuclide for cancer therapy is dependent on many factors (161). Radioconjugates with β (i.e., 131I and 90Y) and α (i.e., 211At and 212Bi) emitters have been proposed for regional therapy of peritoneal implants (162). Estimates of dosimetry suggest that adequate therapy (i.e., 20 Gy) could be delivered to tumors with a depth less than 300 µg using 211At, less than 0.1 cm using 131I, and less than 1 cm using 90Y when conjugated to antibodies. The relatively low radiation dose makes the intraperitoneal delivery route attractive in order to put the source close to the tumor implants in the case of ovarian cancer. Delivery of effective radiation dose is greatly influenced by the extent of tumor binding, depth of tumor penetration, catabolism, and relative distribution between tumor and normal tissues. Antibody-radionuclide conjugates have been successfully developed for non-Hodgkin’s lymphoma resulting in drug approvals with CD20 targeted agents (163). Randomized clinical data in ovarian cancer are sparse with two larger studies having been reported. A randomized trial of 251 patients with Stage 1a or 1b grade 3, or 1c, or 2 ovarian cancer with no macroscopic residual disease were randomized to intraperitoneal radioactive chromic phosphate (32P) versus cisplatin and cyclophosphamide chemotherapy. The cumulative incidence of recurrence at 10 years was 35% (95% CI, 27%–45%) versus 28% (95% CI, 21%–38%) favoring chemotherapy (p = ns). The toxicity profile of 32P was worse (164).
The SMART study was a large randomized study which evaluated intraperitoneal yttrium-90-labeled HMFG1 murine monoclonal antibody in patients with ovarian cancer after a surgically defined complete remission. The MUC-1 antigen that 90Y-muHMFG1 is directed towards is well established as an immunotherapy target and is overexpressed on 90% of adenocarcinomas including that of ovarian cancer. The phase II data supporting the use of 90Y-muHMFG1 administered intraperitoneally in patients in first remission was promising with a Cox model estimate of 10 year survival at 70% compared to 32% of case matched controls (p = 0.003) (165). Patients were stratified using a second surgical assessment where disease status at second look remains a powerful predictor of ultimate progression-free and overall survival (166). Despite the strong preclinical and clinical data supporting this approach, at a median followup of 3.5 years, relapses occurred in 98 versus 104 patients (p = ns). While there is no randomized evidence supporting efficacy of radioconjugates in gynecologic malignancies to date, the number of candidate agents for investigation continues to increase and earlier phase trials are ongoing.
Immunotoxins, Drugs, and Cytokines
Drugs, toxins, and cytokines conjugated to mAbs have been evaluated in numerous cancers. Immunotoxins incorporate an antibody-binding domain and a toxin joined by a chemical cross-linker, peptide, or disulfide bond. The specific targeting afforded by mAbs and the relative potency of the toxin moiety present potential therapeutic advantages. Early conjugates, which have generally not been successful, linked antibodies to standard anticancer drugs such as doxorubicin or vinblastine. It is generally not possible to deliver comparable levels of cytotoxic agents as intravenous administration using this approach. This field is advancing by replacing murine antibodies with humanized versions and conjugating agents that are both specific and functional at lower doses (167). Multiple agents are being evaluated though none have proven efficacy in gynecologic tumors to date.
Other Selected Clinical Trials with Monoclonal Antibodies
Monoclonal Antibodies Targeting CA-125
CA-125, a tumor-specific antigen that is found in 97% of patients with late-stage ovarian cancer, was first described by Bast et al. as an antigen that is elevated in the serum of patients with epithelial ovarian cancer (157). CA-125 has been cloned and identified as the mucin MUC-16 (168), as shown in Figure 15.2. Studies have shown that MUC-16 has a variety of functions and contributes to ovarian cancer cell growth, survival and invasiveness (169). Recent data suggest that expression is extended to digestive tract adenocarcinomas, and moderate expression likewise correlates with poor survival (p < 0.001) (170). The sheer size of the MUC-16 complex has made selecting appropriate sequences for incorporation into vaccines difficult, and initial studies focused on antibodies which could be raised against CA-125.
Monoclonal antibodies including anti-idiotypic antibodies have been used as CA-125 targets. The historical rationale for an anti-idiotype vaccine was an attempt to augment both antibody and T-cell responses by using antibody facsimiles of the native antigens with the goal of overcoming poor immunogenicity. Tumor-associated antigens are traditionally weakly immunogenic as the large majority of them in humans are nonmutated self-antigens. This approach attempted to present the desired epitope to the now tolerant host in a different molecular environment but the problem with many tumor antigens is that they are ill-defined chemically and difficult to purify and produce (171). The “immune network hypothesis” set out to transform epitope structures into idiotypic determinants expressed on the surface of antibodies. It was originally proposed in the 1970s (172
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


