The typical clinical NMS tetrad includes: (1) altered mental status, (2) extrapyramidal symptoms, (3) hyperthermia, and (4) autonomic dysfunction. The early manifestations of the disorder may be subtle, so a high level of suspicion for NMS is required in a patient receiving potentially causative medications who presents with mildly altered mental status and/or mild hyperthermia. Onset is generally within 1 to 7 days of medication initiation or a dose increase, in contrast to the onset of MH and serotonin syndrome which usually have more rapid onset (Table 18.2). NMS is an evolving disorder, and in one review 82% of 340 patients presented with only 1 clinical sign (6). Typically, all of the signs occur within 24 hours and peak in 72 hours. Mental status may range from mild confusion to coma, and encephalopathy precedes other NMS symptoms in the majority of patients (6). The most common extrapyramidal signs are generalized lead-pipe rigidity (joint stiffness that is present throughout the range of passive motion) or dystonia (involuntary persistent abnormal postures of a limb or the torso sometimes with superimposed small repetitive movements), but tremor and myoclonus can also occur. Rarely, NMS may occur without hyperthermia (7). Dysautonomia is common and can include blood pressure lability, tachycardia, tachypnea, and diaphoresis. Patients may also have sialorrhea, incontinence, dysarthria, and dysphagia. As the disorder progresses, a severe hypermetabolic syndrome can ensue, resulting in multi-organ system derangement including cardiorespiratory collapse, renal failure, thromboembolic disease, hyperthermia, and hypoxemic end-organ injury.
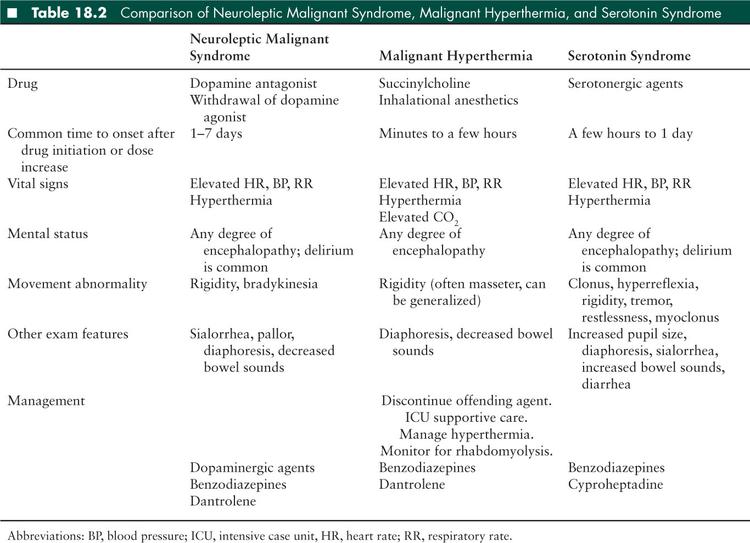
Diagnostic criteria have been proposed (8). Major criteria include hyperthermia, rigidity, and elevated creatine kinase. Minor criteria include tachycardia, abnormal blood pressure, tachypnea, altered consciousness, diaphoresis, and leukocytosis. NMS is very likely if all three major or two major and four minor criteria are present. A recent literature review by a panel of multi-specialty NMS experts used the Delphi process to reach consensus regarding which elements were most important in diagnosing NMS. The panel reached consensus regarding the importance of eight elements in diagnosing NMS: (1) recent dopamine antagonist exposure or dopamine agonist withdrawal, (2) hyperthermia, (3) rigidity, (4) mental status alteration, (5) creatine kinase (CK) elevation, (6) sympathetic nervous system lability, (7) tachycardia plus tachypnea, and (8) a negative workup for other causes. There was consensus regarding critical values for hyperthermia (> 38.0°C on at least two measurements), CK elevation (at least four times the upper limit of normal), hypertension (> 25% above baseline), blood pressure fluctuation (≥ 20 mmHg diastolic or ≥ 25 mmHg systolic change within 24 hours), tachycardia (≥ 25% above baseline), and tachypnea (≥ 50% above baseline). These criteria have not been validated (9).
The differential diagnosis for NMS will sometimes include encephalitis and meningitis, structural brain lesions, sepsis, serotonin syndrome, MH, heat stroke, diencephalic storm, intoxication (anticholinergic agents, MDMA (3,4-methylenedioxy-N-methylamphetamine, ecstasy), amphetamines, phenycyclidine), withdrawal (alcohol, sedatives, L-dopa, amantadine, baclofen), or endocrine abnormalities (thyrotoxicosis or pheochromocytoma). In many instances, testing to exclude other diagnoses will be needed. Malignant catatonia is a serious behavioral syndrome with altered movement including bradykinesia (slow, limited movement) or immobility, mutism, stuporlike state, and many of the features of NMS. In some cases, it cannot be distinguished from NMS except that malignant catatonia can occur in the absence of a provoking drug.
NMS is fatal in about 10% of patients (10). Given the progressive nature and severity of the disorder, all patients with suspected NMS should be admitted to a closely monitored critical care setting. Immediate withdrawal of the antidopaminergic agent and avoidance of further dopamine receptor blockade are required. While there is no controlled trial data regarding NMS management, early intervention and prevention of the more severe systemic problems that result from the hypermetabolic syndrome is important. In some cases, all that is needed is withdrawal of the provoking agent and general support. Treatment is typically individualized based on clinical severity. In mild cases, such as with a low-grade hyperthermia and mild hypokinesis or rigidity, parenteral benzodiazepines such as lorazepam can be administered. If the patient develops more worrisome hyperthermia or extrapyramidal signs (rigidity or dystonia), then a dopamine agonist such as bromocriptine should be administered orally or via a feeding tube. While bromocriptine is often the drug of choice, other dopaminergic agents include carbidopa/levodopa, ropinirole, and pramipexole. If there is moderate hyperthermia and rigidity, then dantrolene (a muscle relaxant that causes excitation-contraction decoupling) can be administered. Dantrolene helps reduce core body temperature by reducing skeletal muscle contraction and hypermetabolism (11). These agents can be coadministered. Treatment is often required for about a week, depending on the half-life of the causative medication. If NMS is caused by a depot injection, then continued treatment may be required for several weeks.
Along with the targeted treatment of the NMS described above, supportive treatment is the mainstay. The goal of supportive treatment is to reduce the damaging effects of the severe hypermetabolism. Adjunctive therapies to maintain normothermia include ice packs, cold intravenous fluids, or surface cooling systems (cooling blankets). Cardiopulmonary monitoring and support is needed, and fluid replacement is important, particularly if rhabdomyolysis occurs. Monitoring for disseminated intravascular coagulation, electrolyte abnormalities, CK elevation, and renal dysfunction is imperative.
If the symptoms of NMS are refractory to pharmacological treatment, particularly if there are prominent catatonic features, electroconvulsive therapy may be used. This may also be a treatment option if there are ongoing catatonic and parkinsonian features after NMS has resolved (11,12).
In patients who recover from NMS, there remains a significant risk of recurrence (up to 30%) if the patient is given a subsequent neuroleptic challenge (3). Starting a neuroleptic after recovery from NMS depends on the degree of need for such a medication and education of the patient about the risks and benefits. If feasible, the patient should not be treated with a neuroleptic for at least several weeks following recovery, drug selection should be based on both efficacy and low incidence of causing NMS, and the drug should be introduced at a low dose and escalated slowly.
■ MALIGNANT HYPERTHERMIA
MH is a life-threatening syndrome that occurs in susceptible patients after depolarizing neuromuscular blockade with succinylcholine or administration of halogenated inhalational anesthetics (halothane, sevoflurane, isoflurane, desflurane). Rarely, MH may occur after exposure to extreme heat or with vigorous exercise. MH typically presents immediately after exposure to the causative medication, but there can be a delay of several hours.
Clinical features of MH involve a hypermetabolic state. A high clinical suspicion is necessary in order to make the diagnosis. A 1994 consensus conference led to the formulation of a set of diagnostic criteria. The more criteria met (especially > 6), the more likely a reaction constitutes MH (13). The criteria included (1) respiratory acidosis (end-tidal CO2 above 55 mmHg or arterial PCO2 above 60 mmHg), (2) cardiac involvement (unexplained sinus tachycardia, ventricular tachycardia, or ventricular fibrillation), (3) metabolic acidosis (base deficit > 8, pH < 7.25), (4) generalized muscle rigidity, (5) rhabdomyolysis (CK > 20,000/L units, myoglobinuria, hyperkalemia), (6) hyperthermia (rapidly increasing temperature, > 38.8°C), (7) other (rapid reversal of MH signs with dantrolene, elevated resting serum CK levels), and (8) a family history of MH (autosomal dominant pattern).
The first manifestation is often a rise in end-tidal CO2 due to muscle hypermetabolism. Other early symptoms include tachycardia and tachypnea in response to catecholamine release and muscle rigidity (often at masseter muscle). The initial presentation may be subtle making diagnosis difficult. Hyperthermia often develops later and rigidity develops in only about half of patients. Rhabdomyolysis, disseminated intravascular coagulation, and cardiac arrest may occur.
The hypermetabolic state and subsequent end-organ dysfunction are similar to what occurs in NMS. A major distinguishing factor between NMS and MH is the exposure itself, but there are some patients who perioperatively receive anesthetic agents as well as dopamine receptor blocking agents, making distinctions more difficult. In MH, the time from the drug exposure to severe hyperthermia and rigidity can be minutes to hours, while the time course for NMS is hours to days (Table 18.2). The differential diagnosis of MH is similar to that of NMS (see above).
MH is a disorder or calcium homeostasis in which calcium accumulation within the sarcolemma causes sustained muscle contraction. MH is an autosomal dominant inherited disorder. The majority of cases (50%–70%) are thought to be caused by a mutation in the ryanodine receptor gene (RYR1). The protein encoded by this gene is located on the sarcoplasmic reticulum and is responsible for excitation-contraction coupling. With abnormal RYR1 protein, calcium release is excessive and sustained skeletal muscle contraction ensues (14). Patients with MH may describe a family history of similar reactions. RYR genetic testing is available, but since there is heterogeneity a normal test does not prove absence of MH. An in vitro contracture test has been developed, in which the response of a muscle fiber to halothane or caffeine is investigated, but this requires a surgical procedure and is not widely available.
The mortality of untreated MH is high, but it is less than 5% with modern pharmacological intervention and supportive care (15). Guidelines for management are available from the malignant hyperthermia society (www.MHAUS.org). The provoking agent must be discontinued immediately. If the patient is still in the operating room then intravenous anesthesia should be initiated. Close observation in an intensive care unit (ICU) is required. One hundred percent oxygen should be administered. Ventilation rate may be increased to normalize PaCO2. Sedation using benzodiazepines is often required. Body cooling may be required involving ice packs, cold intravenous fluids, or surface cooling system (cooling blankets). Rhabdomyolysis management involves fluid hydration to maintain a high urine output. If pharmacologic paralysis is required for intractable rhabdomyolysis or hyperthermia, then nondepolarizing neuromuscular blocking medications should be used such as vecuronium. Monitoring for signs of disseminated intravascular coagulation is important. Potassium levels may rise. Parenteral dantrolene is the only specific treatment for MH, and it should be initiated immediately upon recognition of the disorder (16). Dantrolene 2.5 mg is given by intravenous bolus (17). Since the introduction of dantrolene, mortality related to MH has decreased. Dantrolene antagonizes RYR-mediated calcium release. Guidelines addressing the transfer of patients from ambulatory surgical centers to higher level facilities should MH occur recommend dantrolene administration prior to transfer (18). Recurrence may occur, usually within 1 day of the initial reaction, so prolonged monitoring well past the start of improvement is important.
Some patients with myopathy may have an increased risk for MH (19). These include central core disease, multiminicore disease, nemaline rod myopathy, a sodium channel form of myotonia, or hypokalemic periodic paralysis. Central core disease is the most linked to MH and is a congenital myopathy with autosomal dominant transmission related to glycolytic enzyme deficiency that presents with generalized weakness in early infancy.
MH diagnosis has implications for both the patient and family members. Patients and their families must be informed of the diagnosis and implications, and the drug reaction should clearly be listed in the permanent medical record since it impacts future anesthetic use. Many patients with known MH risk wear a medical alert bracelet. If anesthesia is needed, then agents should include nondepolarizing neuromuscular blocking agents (such as vecuronium and rocuronium), nitrous oxide, and intravenous anesthetics (benzodiazepines, barbiturates, propofol, thiopental, ketamine, etomidate).
■ SEROTONIN SYNDROME
Serotonin syndrome has also been referred to as serotonin toxicity, serotonin behavioral syndrome, and serotonin hyperactivity syndrome. This toxidrome was initially described in 1960 in patients who were using monoamine oxidase inhibitors (MAOIs) and tryptophan concurrently (20). It is caused by medications that augment serotonin transmission. It can occur when a single serotonergic medication is used at an appropriate therapeutic dose, but it is more common when an overdose occurs (19) or when two or more serotonergic medications are used simultaneously. Combined use should be avoided, but if necessary, the two drugs should be kept at as low dose as feasible. The patient must be particularly well-educated about recognizing and reporting toxicity symptoms. Complete documentation of prescribed and over-the-counter medications and knowledge of medication effects with careful prescribing practices will minimize the risk of this sometimes fatal disorder. Table 18.3 is a list of medications that affect serotonin metabolism. There are various mechanisms by which serotonin activity can be increased in the central nervous system. These include direct serotonin agonists, inhibitors of serotonin reuptake from the synaptic terminal, serotonin release augmenting medications, and medications that prevent serotonin degradation. Dopamine agonists are also implicated in serotonin syndrome; it is thought that increased concentrations of dopamine in the central nervous system contribute to serotonin release. Note that Table 18.3 includes illicit drugs. This adds to the need for obtaining a complete history.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


