Hormones and Human Malignancies
G. LARRY MAXWELL  MIAN M. K. SHAHZAD
MIAN M. K. SHAHZAD  LAUREL W. RICE
LAUREL W. RICE
Hormones have been linked to several of the most commonly occurring human malignancies, including breast, endometrium, ovary, and colon. The ability of hormones to stimulate cellular proliferation, leading to random genetic errors, is assumed to be the basis by which these peptides promote the development of human neoplasia. The pathways and mechanisms by which hormones affect their biologic responses is an exploding area of research, largely as a result of the tremendous potential for translational research and improved patient care. This chapter reviews the relationship between hormones and human malignancies, specifically as they relate to risk, prevention, and treatment.
HORMONE RECEPTOR PATHWAYS AND MECHANISMS
Steroid hormones perform critical functions in morphogenesis and normal physiology of female reproductive organs (1). These hormones are involved in many growth and developmental processes and exert their physiologic effects by binding to their particular receptors (2). Steroid hormone receptors are part of the ligand-activated nuclear transcription factor superfamily. The members of this superfamily also include glucocorticoid, mineralocorticoid, androgen, thyroid, retinoid, progesterone receptors (PR), and estrogen receptors (ER) (3). In the inactive state, these receptors are bound to repressor/chaperone complexes within the cytoplasm. Upon binding to their specific ligands, the receptor-ligand complex undergoes dimerization, which is followed by nuclear localization (leading to the name “nuclear receptor”). Inside the nucleus, the steroid hormone–receptor complex binds to specific DNA sequences called response elements. (4). Also participating in this interaction are nuclear receptor coactivators and corepressors, as well as the general transcription machinery (5). The net result of these interactions is the transcriptional activation or repression of hormone-responsive genes. More recently, nongenomic actions of ER and other nuclear receptors have been documented. These effects result from protein-protein interactions and membrane-associated nuclear receptor action (6).
Steroid Hormone Biosynthesis
While a comprehensive account of steroidogenesis is beyond the scope of this chapter, a succinct synopsis is presented (Fig. 16.1) to underscore targets of therapeutic relevance. All steroid hormones are derived from cholesterol. In steroid biosynthesis, cytochrome P450-linked side-chain cleaving enzyme (P450 ssc) acutely regulates the rate-limiting step and converts cholesterol to pregnenolone (7). This naturally produced hormone in the body is sometimes referred to as the “mother hormone” and serves as a precursor for dehydroepiandrosterone (DHEA) and all steroid and sex hormones. Conversion of cholesterol to pregnanolone in the ovary is tightly regulated by follicle-stimulating hormone (FSH) and luteinizing hormone (LH) via cAMP and protein kinase A (PKA) signaling (8). This leads to increased production of androgen intermediates, androstenedione and testosterone, that are then converted by the P450-aromatase enzyme to estrone and estradiol (E2), respectively. P450-aromatase is not exclusively expressed in ovarian tissues. Interestingly, its expression in many other tissues results in biosynthesis of local E2 (9). The availability of locally produced E2 may be particularly important in both men and postmenopausal women, where ovarian hormone production is lacking. High levels of locally synthesized and readily available E2 have also been implicated in breast carcinogenesis (10).
Recently, Ghosh et al. have solved the crystal structure of CYP19A1 and revealed a small active cleft that is well suited to bind androstenedione snugly, prior to its conversion to estrone (11). Data also indicate that CYP19A1 has wide tissue distribution (including placenta, ovary, testes, and adipose tissue), and its expression has been demonstrated to be associated with about 75% of estrogen-dependent breast cancers (12). Aromatase inhibitors in this specific patient population have resulted in a substantial reduction in the risk of breast cancer recurrence (13). Although other risks emerge, including grade 3 and 4 cardiovascular events and bone fractures (14,15). Additionally, we now know that several tissue types have tissue-specific expression of the CYP19A1 gene promoter, and the presence of tissue-specific promoters has raised the possibility of developing more selective aromatase inhibitors. Such agents would be able to, for instance, block E2 in breast cancer, without affecting estrogenic pathways in bones (16,17).
Steroid Hormone Receptor Structure
Steroid receptors are comprised of six functional domains (Fig. 16.2) (18). The A/B domain is highly variable among the different members of this family and contains within it the hormone-independent activating 1, or AF-1, domain. The relatively conserved Cregion contains the DNA-binding domain (DBD). Within the central DBD, two zinc fingers target the receptors to their corresponding hormone response elements. The DBD binds as a dimer, with each monomer recognizing half of the palindromic DNA sequence of the response element. The D domain is a peptide linker that connects the DBD to the ligand-binding domain (LBD). The E region is a multifunctional domain and contains amino acids involved in ligand binding, receptor dimerization, nuclear localization, coactivator/corepressor interaction, and ligand-dependent activating function (AF-2). The F region is a variable extension of the E region.
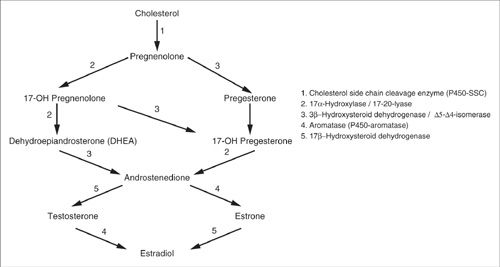
FIGURE 16.1. Molecular biology of steroid hormone synthesis. Notice the important role of aromatase in the conversion of androgens to estradiols.
Estrogen Receptors
The ER mainly functions as a ligand-regulated, DNA-binding transcription factor, resulting in modulation of gene expression of proteins involved in key pathways related to cancer progression. However, additional ER functions, independent of DNA binding, have also been reported (19,20). ERs can be recruited to the DNA by indirect tethering through other DNA-bound transcription factors, including members of the activating protein-1 (AP-1) transcription family (4,21,22). Due to alternative RNA splicing, several isoforms of the ER are known to exist, but ER-α and ER-β are considered to be the main isoforms. We now know that ER-α is encoded by the ESR1 gene and ER-β is encoded by the ESR2 gene. The ESR1 and ESR2 genes are located on the 6th and 14th chromosomes (6q25.1 and 14q23), respectively (23).
In humans, the ERs exhibit 96% and 58% homology in their DBD and LBD, respectively. These structural differences in ERs lead to discrete ligand affinities and physiological properties. For example, estrone and raloxifene bind preferentially to ER-α, while estriol and genistein have higher binding affinity for ER-β. Furthermore, tamoxifen and raloxifene exhibit partial agonist activities after binding to ER-α, while they act as pure antagonists when bound to ER-β (24). This is thought to result from distinct conformational changes induced by ligand binding in the two receptors (25).
Both ERs are widely expressed in many tissue types; however, there are important differences in the pattern of their expression. ER-α is the predominant ER isoform in many tissues, including breast cancer, endometrium, ovarian stroma, and the hypothalamus. ER-β has notably higher expression in ovarian granulosa cells, prostate gland, gastrointestinal tract, endothelial cells, heart, lung, kidney, brain, and bone (26–28). However, some expression of both receptors is observed in many tissues. Data now indicate a correlation between ER-β transcript levels and expression of oncogenes in cancer cells and also suggest a role of ER-β in endometrial carcinogenesis (29). As noted above, ER-α and ER-β have been shown to have unique physiologic properties. In addition, they are capable of forming ER-a (aa) or ER-b (bb) homodimers or (ER-α/ER-β) heterodimers, thus influencing each other’s function (30). In this context, ER-β has been shown to function as a dominant inhibitor of ER-α (21). Therefore, it is likely that normal physiologic function in E2 responsive tissues relies on the relative expression ratio of ER-α and ER-β in those tissues. Recently, Iyer and colleagues have shown that estrogen promotes ER-negative breast tumor growth by recruitment of pro-angiogenic bone marrow-derived myeloid cells in murine xenograft models, and the researchers demonstrated that ER-α expression is present only in bone marrow-derived cells and this expression was required and sufficient to promote breast tumor growth when challenged with estrogen (31).
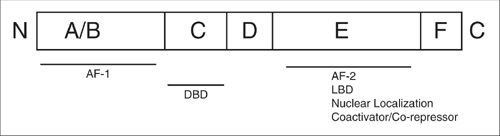
FIGURE 16.2. Functional domains of the human estrogen receptor. The A/B domain is highly variable among the different members of this family and contains the hormone-independent activating 1, or AF-1, domain. The C region consists of the DNA-binding domain (DBD). The D domain is a peptide linker that connects the DBD to the ligand-binding domain (LBD). The E region is a multifunctional domain and contains the LBD as well as amino acids involved in receptor dimerization, nuclear localization, coactivator/corepressor interaction, and ligand-dependent activating function (AF-2). The F region is a variable extension of the E region.
Accumulating evidence suggests that changes in normal ER-α-to-ER-β ratio may be associated with neoplastic states. Cancers of the ovary, colon, and endometrium are all characterized by a relative loss of the normally predominant ER subtype when compared with normal tissue (32–36). Whether these changes predispose to or are a result of carcinogenesis remains to be elucidated. The development of ER-α, ER-β, and double knockout mice (ERKO, βERKO, and αβERKO, respectively) has helped with the identification of each receptor’s physiologic role in its target organs (37). These findings are summarized in Table 16.1. Recently, genotyping data from more than 12,000 BRCA1 and 7,000 BRCA2 mutation carriers compiled from 40 studies identified ER-related SNP (rs67397200) at chromosome 19p13.1 that portends an increased risk of ovarian cancer for BRCA1 (HR 1.16) and BRCA2 (HR 1.30) mutation carriers. Identification of this cancer susceptibility locus may also be useful for ovarian cancer risk prediction for BRCA1 and BRCA2 mutation carriers (38).
Several of the ER-α and ER-β “splice variants” have been demonstrated to possess altered hormone binding and/or transcriptional properties. Some can act in a dominant negative fashion by dimerizing with the wild-type receptors and interfering with their function in vitro (39). As mentioned above, ER has been shown to regulate transcription by interacting with other transcription factors and signal mediators (40–42). This mechanism involves protein-protein interaction and does not require DNA binding. An example of this type of interaction is the influence of ERs on the transcriptional activity mediated by the components of the AP-1 pathway such as jun and fos (34). In the presence of ER-α, E2 stimulates AP-1–dependent transcription, while tamoxifen and raloxifene inhibit it. In contrast, ER-β produces the opposite effect, with estradiol inhibiting and tamoxifen and raloxifene stimulating AP-1–mediated transcription. The activation of ER-α also results in induction of fos-related antigen 1 (fra-1) and regulates uterine stromal differentiation and remodeling during early pregnancy (43). An additional mechanism of ER action involves “cross-talk” with growth factors including insulin-like growth factor 1 (IGF-1), epidermal growth factor (EGF), and transforming growth factor β (TGF-β) (44,45). EGF can mimic the effects of E2 on the reproductive tract. This effect is dependent on ER-α because it can be blocked by ER antagonists and is absent in ERKO mice, despite an intact EGF signaling cascade (46). Mechanisms for this cross-talk involve signal transduction from growth factor receptors via the ras/raf/MAPK, as well as the AKT/PI3K cascade intermediaries culminating in the phosphorylation and activation of ER’s AF-1 domain (47). AKT activation results in cell survival and inhibition of apoptosis. In normal tissue, this pathway is kept in check by a phosphatase called PTEN. PTEN is a tumor suppressor that negatively regulates the PI3K/AKT pathway by dephosphorylating PIP3. The activation of the PI3K/AKT pathway is particularly relevant in endometrial adenocarcinoma, where up to 80% of malignancies and 55% of premalignant lesions have been shown to harbor PTEN mutations (48,49). Most investigations of the growth factor–ER cross-talk have involved ER-α. However, positive regulation of AKT over ER-β was recently reported via phosphorylation of glucocorticoid receptor-interacting protein 1 (GRIP1) transcriptional coactivator (50).
For decades it has been observed that exposure to estrogens can lead to immediate physiologic sequelae independent of transcriptional effects. These observations supported the presence of ERs outside the nucleus as the mediators of these nongenomic steroid hormone effects (4,51). The nongenomic effects of estrogens appear to follow at least two major pathways. The interactions of ER-α and its splice variants with components of the growth factor signaling cascades including MAPK and PI3K pathways are important (52,53). Recently, in vitro and in vivo data indicate that estrogen promotes ovarian cancer growth both directly and indirectly, by making the tumor microenvironment more conducive to tumor growth (54). Here it is essential to recognize that cellular exposure to estrogen results in a multitude of functions that can be measured with distinct kinetics. Specifically, the rapid events that take place within minutes are termed pre-genomic and utilize the second messenger pathway. The pre-genomic events may also ultimately lead to changes in gene transcription, as do the delayed events (55). This mechanism involves a G-protein coupled receptor, GPR30, an intracellular transmembrane receptor that localizes mainly to the endoplasmic reticulum. In this pathway, estrogens diffuse through the plasma membrane and bind GPR30 at the endoplasmic reticulum. This results in intracellular calcium mobilization and synthesis of phosphatidylinositol 3,4,5-trisphosphate in the nucleus. Activation of this pathway has been demonstrated in cells that lack ER-α ER-β expression (56).
Summary of Findings from Estrogen Receptor Knockout Mice Experiments |
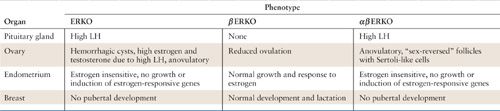
Abbreviation: LH, luteinizing hormone.
Progesterone Receptors
The progesterone receptor (PR) is an intracellular steroid receptor that belongs to the nuclear receptor subfamily of receptors (i.e., NR3C3) (57). These nuclear hormone receptors, in general, are involved in diverse physiological functions, including differentiation, embryonic development, and cellular homeostasis. However, dysregulation of nuclear receptor superfamily members has been implicated in many disease states, including cancer (58). Multiple mechanisms through which PR mediates gene expression are regulated by numerous cellular processes that originate either within the cell or in the extracellular environment (59). The effects of progesterone are mediated by two PR proteins, PRA and PRB. In humans, the two PR proteins are encoded by the gene, PGR, which is located on chromosome 11q22 (60). These 2 receptors differ due to the additional 164 amino acids present in the N-terminus of PRB. Both PRA and PRB contain transcription-activating functional (AF) domains: AF-1 and AF-2. The N-terminal region of PRB contains an additional AF domain (AF-3) (61). The two PRs exhibit differences in function that are cell context and promoter dependent. For example, PRB uniformly exhibits progestin-dependent transactivation in the various cell types examined. However, PRA’s transcriptional activity is cell and response-element specific. When tested on simple progestin response elements (PRE), PRA and PRB display similar transactivational activity. However, PRA’s activity is reduced or eliminated on more complex response elements (62). In addition, PRA has been shown to act in a dominant negative fashion, antagonizing the transcriptional activity of not only PRB, but also other nuclear receptors such as the glucocorticoid, mineralocorticoid, androgen, and estrogen receptors (63).
In general, most tissues express equal levels of the two PR isoforms. However, differential expression can be observed, as each promoter is regulated independently. For example, in the endometrium, stromal cells express predominantly PRA throughout the menstrual cycle, whereas the epithelial cells switch from PRA to PRB during the early secretory phase (64). PRA knockout mice (PRAKO) exhibit endometrial hyperplasia mediated by PRB following exposure to progestins (65). Similar studies using PRB knockout mice (PRBKO) have shown that PRA is sufficient for eliciting progesterone’s reproductive responses in the ovary and the uterus. In contrast, PRB is required to elicit normal proliferative responses of the mammary gland to progesterone (66). Finally, changes in PR isoform relative or overall expression have been described in endometrial, ovarian, and breast cancers (30,46,67).
It is generally thought that the expression of PR in ovarian cancer may be protective (68,69). Recent data indicate that PRB activation by other second messengers (e.g., cAMP) results in cell cycle arrest, induction of cellular senescence, and suppression of oncogenicity of ovarian cancer (70). A decrease in PGR content due to loss of heterozygosity and/or polymorphisms of PGR are associated with increased risk of ovarian cancer, and signifies that activation of PGR may guard against the development of ovarian cancer (71–73). Preclinical data now indicate that certain microRNAs may also regulate the expression of PGR, and targeting of PGR by specific microRNAs is seen in transition from normal to endometrial cancer states (74). Additional evidence supports that progesterone may also signal through other receptors that may be localized to the membrane (75). For example, Peluso and colleagues, using orthotopic ovarian cancer mouse models, recently demonstrated that progesterone membrane component-1 regulates the development and sensitivity to cisplatin in ovarian cancer (76).
Nongenomic functions of PGR involve the extranuclear PGRs that associate through proline-rich domain to interact with SH3 domain of Src kinase. This complex cascade involves activation of the EGFR/c-Src/Ras/ERK pathway and the PI3K/AKT pathway, resulting in a pro-tumorigenic role in cancer cell biology (77,78,79). More recently, progesterone was shown to mediate mammary gland stem cell self-renewal (80). Knowing that PGR function is altered by the action of protein kinases and that activation of PGR can lead to the activation of several kinases known to enhance tumor progression (81), it is essential to understand these critical molecular interactions in depth to significantly improve our approaches for prevention and treatment of human malignancy.
HORMONES AS RISK FACTORS FOR HUMAN MALIGNANCIES
Exogenous Hormones and Modulation of Cancer Risk
Breast
Hormone Replacement Therapy
Women without history of breast cancer. Multiple epidemiologic investigations have evaluated the relationship between hormone replacement therapy (HRT) and breast cancer. In a meta-analysis of 51 studies of 52,705 women with breast cancer and 108,411 women without breast cancer, the Collaborative Group on Hormonal Factors in Breast Cancer (CGHFBC) determined that the risk of developing breast cancer is increased in women using HRT. The risk increases especially among women with >5 years of use (relative risk [RR], 1.3; 95% confidence interval [CI], 1.21–1.49). The cumulative incidence of breast cancer was 45 per 1,000 women between the ages of 50 and 70 years, and use of HRT for >5 years was associated with an estimated cumulative excess of 2 breast cancers for every 1,000 users. The increased risk of breast cancer associated with prolonged HRT was greater for women with low weight or low Body Mass Index (BMI). Breast cancers diagnosed in women who had a history of HRT was inclined to be less advanced clinically compared with women with a negative history of HRT use (82). The Breast Cancer Detection Demonstration Project, a cohort study with 46,355 postmenopausal women, 2,082 of whom were diagnosed with incident cases of breast cancer, reported that women who had taken unopposed E2 had a 1.2-fold (95% CI, 1.0–1.4) increase in breast cancer risk, while women taking combination E2 and progestin regimens had a 1.4-fold (95% CI, 1.1–1.8) increased risk. Breast cancer risk was significantly elevated only in recent users with a BMI less than 24 (83). Finally, a recently reported case-control study involving 1,897 women with breast cancer revealed that women using combination HRT had a statistically higher risk of breast cancer (odds ratio [OR], 1.24; 95% CI, 1.07–1.45) than women taking unopposed E2 (OR, 1.06; 95% CI, 0.97–1.15) (84).
The Women’s Health Initiative (WHI), a randomized, double-blind, placebo-controlled trial, recently reported the interim analysis of 8,506 women who received continuous combination HRT (0.625 mg of conjugated equine estrogen [CEE] plus 2.5 mg of medroxyprogesterone acetate [MPA]) versus 8,102 women prescribed placebo. The Data and Safety Monitoring Board for the trial recommended early cessation of this trial arm secondary to the observed adverse effects. Interim results from the WHI study revealed that the risk of coronary heart disease (hazard ratio [HR], 1.29; 95% CI, 1.02–1.63), stroke (HR, 1.41; 95% CI, 1.07–1.85), and pulmonary embolism (HR, 2.13; 95% CI, 1.39–3.25) were all significantly elevated in patients receiving continuous combination HRT. The observed increased risk of breast cancer (HR, 1.26; 95% CI, 1.0–1.59) was similar to results from the aforementioned observational studies (85). A more detailed evaluation of the WHI data used a weighted Cox proportion hazards analysis in an effort to account for the lag in time required to achieve the full effects of the hormone on breast cancer incidence. In this analysis, the rate of invasive breast cancer was increased in patients receiving progestin plus estrogen (HR, 1.24, weighted p < 0.003) (86). Extended follow-up of the patients from the WHI intervention trial was continued among 8,052 surviving participants (95%) in a post-intervention study for a mean of 8 years. More breast cancers were diagnosed among women receiving CEE plus MPA versus placebo (HR, = 1.27; 95% CI, 1.06–1.51) (87). Estrogen plus MPA had a greater influence on breast cancer incidence than placebo if initiated closer to menopause (88). The breast cancer developing in patients receiving the hormonal regimen was also not restricted to hormone receptor–positive tumors (89).
Further analysis has revealed that breast cancer diagnosed in women receiving CEE plus MPA, when compared with placebo patients diagnosed with breast cancer, were more likely to have metastatic node involvement, even though between the two groups the tumors were similar in terms of grade and histology (88). The combination of CEE plus MPA compared with placebo has been shown to interfere with mammographic detection, potentially leading to breast cancers being diagnosed at a later stage of disease (90). Moreover, a 11-year follow-up analysis recently has shown that breast cancer mortality has increased among women who received CEE plus MPA compared with placebo (88). Since the original results from the WHI were released in 2002, there has been a decrease in the nationwide use of postmenopausal hormone therapy (91). In addition, patients on HRT were diagnosed with larger tumors at a more advanced stage, compared with patients who received placebo (88).
In the WHI trial, 10,739 postmenopausal women with prior hysterectomy received 0.625 mg of CEE (n = 5,310) versus placebo (n = 5,429). In the initial manuscript, the WHI investigators reported that there was a possible reduction in breast cancer incidence, but the results were not statistically significant (HR, 0.77; 95% CI, 0.59–1.01) (92). Further analysis of the data revealed that treatment with CEE for 7 years does not appear to increase the incidence of breast cancer (77). Extended follow-up of the patients from the WHI intervention trial was continued among 7,645 surviving participants (78%) in a post-intervention study for a mean of 10 years. Analysis of intervention and post-intervention data revealed a lower incidence of breast cancer among patients receiving CEE (HR, 0.77; 95% CI, 0.62–0.95) (93).
Women with a history of breast cancer. Physicians are apprehensive about prescribing HRT to women with a positive personal history of breast cancer. The theoretical concern has been that exogenous hormones may stimulate the growth of dormant microscopic disease and ultimately lead to a decreased disease-free interval and survival.
Several case-control studies have reported that the rates of breast cancer recurrence among survivors taking HRT vary from 3% to 7% (94,80) and do not appear to significantly diverge from stage-specific population rates of recurrence. In a case control study by DiSaia et al. (81), 41 sporadic breast cancer patients taking HRT were matched to controls according to age, stage, and socioeconomic status. A majority of the patients had early-stage disease. Recurrences were detected in 12 patients with stage I disease, 1 with stage II, and 2 with stage III. No evidence of increased disease recurrence was noted among the patients receiving HRT. In a subsequent analysis by the same authors, 125 sporadic breast cancer survivors receiving HRT were compared with 363 control subjects matched according to age at diagnosis, stage of breast cancer, and year of diagnosis. The analysis revealed that the risk of death was lower among the breast cancer patients receiving HRT (OR, 0.28; 95% CI, 0.11–0.71). According to the American College of Obstetricians and Gynecologists, the projected absolute lifetime risk for non-HRT users is approximately 10 cases of breast cancer per 100 women. This risk would increase to 12 cases per 100 women using unopposed E2 and 14 cases per 100 women using combined E2 and progestin (95). Data from a prospective, randomized trial is not available to answer this question.
Oral Contraceptives
Since their introduction in the 1960s, oral contraceptives (OCPs) have been taken by over 200 million women throughout the world. The CGHFBC performed a meta-analysis of 54 epidemiologic studies on 53,297 women with breast cancer and 100,239 women without breast cancer. Women who were current users or had used OCPs in the previous 10 years were at an increased risk of breast cancer (RR, 1.24; 95% CI, 1.15–1.33). Among women taking OCPs, the breast cancers that developed were less advanced than those diagnosed in women with no history of OCP use (96). Because the results from this meta-analysis reflected data from studies completed over the prior 25 years, the Women’s Contraceptive and Reproductive Experiences (Women’s CARE) study was initiated in an effort to provide a more contemporary analysis of the relationship between OCPs and breast cancer risk (97). In this case-control study, a total of 4,574 women with breast cancer and 4,682 controls were compared. The RR was 1.0 (95% CI, 0.8–1.3) for current OCP use and 0.9 (95% CI, 0.8–1.0) for previous use, suggesting that among women aged 35 to 64 years, current or former OCP use is not associated with a significantly increased risk of breast cancer.
Women with BRCA mutations. In the meta-analysis by the CGHFBC, the increased risk of breast cancer associated with OCP use was not influenced by a family history of breast cancer (96). However, a threefold increased risk of breast cancer was observed among women taking OCPs who had a positive family history of breast cancer. This increased risk was observed only in women using high potency estrogen formulations before 1975 (98). A subsequent matched case-control study by Narod et al. revealed that among BRCA1 carriers, an increased risk of early-onset breast cancer was evident in women who used OCPs prior to 1975 (OR, 1.42; 95% CI, 1.17–1.75), used OCPs prior to age 30 (OR, 1.29; 95% CI, 1.09–1.52), or who used OCPs for 5 or more years. Despite the increased risk associated with OCP usage among BRCA1 carriers, an increased risk of breast cancer was not observed among women with a BRCA2 carrier status (99). In BRCA1 carriers, the protective effects of OCPs against ovarian cancer must be weighed against the possible increased risk of OCP use and breast cancer.
Ovary
Hormone Replacement Therapy
Women without a history of ovarian cancer. Recently, several well-designed case-control and cohort studies have evaluated the relationship between HRT and ovarian carcinoma. The American Cancer Society’s Cancer Prevention Study II found that after accounting for OCP use and parity, postmenopausal women using HRT had higher rates of ovarian cancer-related deaths compared with nonusers (RR, 1.51; 95% CI, 1.16–1.96). The increased mortality risk was most notable for women using HRT for 10 or more years and persisted for up to 29 years after HRT cessation (100). This study is limited by the lack of information regarding estrogen and progestin potency. Another cohort study of 44,241 former participants in the Breast Cancer Detection Demonstration Project identified 329 women who developed ovarian cancer during follow-up. Multivariant analysis revealed an increased association of estrogen-only HRT with ovarian cancer (RR, 1.6; 95% CI, 1.2–2.0) after accounting for age, menopause type, and OCP use (101). In contrast, an increased risk of ovarian cancer was not noted among women who used estrogen and progestin. A Swedish case-controlled trial evaluated 655 case subjects with ovarian cancer and 3,819 control subjects. An increased risk of ovarian cancer was noted among women with a history of either estrogen (OR, 1.43; 95% CI, 1.02–2.00) or estrogen with sequentially added progestins (OR 1.54; 95% CI, 1.15–2.05), particularly with hormone use exceeding 10 years. However, use of estrogen with continuously added progestin was not associated with an increased risk of ovarian cancer when compared with nonuse (102). The latter 2 studies suggest that HRT regimens containing both E2 and progestin may not have the increased risk of ovarian cancer observed with E2 therapy alone. However, the WHI recently identified, in women taking continuous combined HRT, an HR of 1.58 (95% CI, 0.77–3.24) for developing invasive ovarian carcinoma (103). The importance of the presence or absence of progesterone in HRT, as well as the type and potency, remains to be elucidated.
Women with a history of ovarian cancer. Only one randomized, controlled clinical trial exists evaluating the risk of ovarian cancer recurrence in patients receiving HRT. This study was designed to detect a 20% difference in survival between the two treatment groups. In this study, 130 patients less than 59 years of age with invasive ovarian cancer were randomized to continuous E2 HRT or no supplementation. The median disease-free interval for women receiving HRT versus those not receiving HRT was 34 months and 27 months, respectively, not reaching statistical significance (104). Multiple epidemiologic studies have revealed that OCPs are not a risk factor for ovarian cancer, but rather a means of prevention. See below.
Endometrium
Hormone Replacement Therapy
Women without a history of endometrial cancer. Multiple case-control and cohort studies also have suggested that the risk of endometrial cancer is elevated among patients receiving unopposed estrogen. A meta-analysis of 30 studies indicated that the summary RR of endometrial cancer was 2.7 for patients receiving unopposed estrogen (105). The use of unopposed estrogen has also been shown to be associated with the development of endometrial hyperplasia. In the Postmenopausal E2/Progestin Intervention (PEPI) trial, 875 patients were randomized to receive one of five regimens: (a) placebo; (b) CEE, 0.625 mg/day; (c) CEE, 0.625 mg/day plus continuous MPA, 2.5 mg/day; (d) CEE, 0.625 mg/day plus MPA, 10 mg for 12 days each month; or (e) CEE, 0.625 mg/day plus micronized progesterone (MP), 200 mg/day for 12 days per month. Patients receiving unopposed estrogen had a significantly increased risk of atypical adenomatous endometrial hyperplasia (34% vs. 1%) (106).
The addition of progestin to HRT has been shown to eliminate the increased risk of hyperplasia associated with unopposed E2. Although progestins have been associated with the effective treatment of endometrial hyperplasia, the data supporting the protective effects of progestins in HRT are limited. Many of the initial HRT regimens combining progestin and estrogens utilized the progestin for 7 days a month (107). Subsequent studies have recommended prolongation of the progestin component to at least 10 days each month. A case-control study of 832 endometrial cancer patients and 1,114 controls in Washington State revealed that patients using a regimen of estrogen combined with progestin (0.625 mg/day CEE, 2.5 mg/day MPA) had a significantly lower RR (RR, 1.4; 95% CI, 1.0–1.9) of endometrial cancer compared with patients who had taken unopposed estrogen (RR, 4.0; 95% CI, 3.2–5.1). However, patients receiving less than 10 days of cyclic progestin had an RR that was much higher (RR, 3.1; 95% CI, 1.7–5.7) than patients receiving 10 to 21 days of cyclic progestin (RR, 1.3; 95% CI, 0.8–2.2). Patients using either regimen of cyclic combination HRT had an increased risk of endometrial cancer when these regimens were taken for a duration exceeding 5 years (cyclic progestin <10 days per month: RR 3.7; 95% CI, 1.7–8.2; cyclic progestin 10 to 21 days each month: RR, 2.5; 95% CI, 1.1–5.5) (108). Subsequent data from the same investigators suggested that use of a continuous progestin HRT regimen may actually be protective against endometrial cancer (109). The largest case-control study of continuous HRT involved 79 case subjects and 88 control subjects, and it revealed that the RR of patients receiving continuous HRT was 1.07 (95% CI, 0.80–1.43) (92). Data from the WHI indicated that the HR for endometrioid endometrial cancer (EEC) was 0.81 (95% CI, 0.48–1.36) in patients receiving continuous HRT with average follow-up of 5.6 years (103). Extended follow-up of the patients from the WHI intervention and post-intervention trials over 8 years revealed that the incidence of endometrial cancer was still decreased among patients receiving CEE plus MPA compared with placebo, but this observation was not statistically significant (HR, 0.78, 0.52–1.16) (87).
There has recently been some suggestion that the degree of endometrial cancer risk reduction may be dependent on the type of progestin used in the continuous HRT regimen. A Swedish case-control study found that patients with a continuous HRT involving testosterone-derived progestins (i.e., norhisterone, norhisterone acetate, levonorgestrel, lynestrenol) were associated with a lower RR of endometrial cancer than those patients whose regimens involved progesterone-derived progestins (i.e., medroxyprogesterone) (110). Data from the Continuous Hormones as Replacement Therapy (CHART) study found no evidence of endometrial hyperplasia in participants who received norethindrone acetate (a testosterone-derived progestin). In this clinical trial, 1,265 patients were randomized to placebo versus one of eight treatment groups involving either various doses of unopposed ethinyl estradiol or various combinations of ethinyl estradiol plus norethindrone. A total of 1,134 endometrial biopsies were performed over a 2-year follow-up period among women who received the combination HRT regimen; 1,232 biopsies were performed in the women given unopposed E2. As the dose of unopposed ethinyl estradiol was increased, there were increased percentages of subjects with endometrial hyperplasia. There were no cases of hyperplasia noted among the subjects who received different doses of the testosterone-derived progestin norethindrone. The protective effect was noted even among patients randomized to receive doses of norethindrone as low as 0.2 mg (106). In contrast, the PEPI trial identified hyperplasia by a surveillance endometrial biopsy in 3 of 339 patients who received E2 with a progesterone-derived progestin. Further studies are needed to determine the degree of endometrial cancer risk influenced by the progestational activity of the progestin.
Women with a history of endometrial cancer. The use of E2 among patients with endometrial cancer is controversial. In vitro treatment of Ishikawa endometrial cancer cells with estrogen upregulates ER expression and augments growth in endometrial cancer cells (111,112). However, in vivo growth of residual microscopic endometrial cancer in patients has not been proven to date. Four retrospective cohort studies have evaluated patients with early-stage endometrial cancer receiving E2 postoperatively. Creasman et al. evaluated 221 patients with stage I disease, of whom 47 received postoperative HRT and 174 did not (113). Patients receiving HRT were followed for a median of 26 months. Regression analysis revealed that there was no increased risk of recurrent disease associated with HRT use, even when adjusting for age, tumor grade, myometrial lymph node status, and peritoneal cytology. A subsequent study by Lee et al. (114) compared the outcomes of 44 low-risk endometrial cancer patients (stage IA/IB G1/G2 disease) who received HRT following treatment, with 62 patients who received no HRT. There were no recurrences observed among the endometrial cancer patients receiving HRT for a median duration of 64 months. Investigators from the University of California, Irvine, have also failed to demonstrate an increased risk of recurrent disease associated with postoperative HRT (115).
In the first study, investigators evaluated 123 women with surgical stage I and II endometrial cancer. Sixty-one women received postoperative estrogen, and the mean duration of follow-up was 40 months. The disease-free interval was not significantly shortened among the patients receiving E2 (116). The second study was a retrospective cohort study that involved 75 patients with stage I–III disease who received HRT (51% E2 only, 49% E2 with added progestin) postoperatively. This group of patients was then matched according to decade of age at diagnosis and stage of disease to a group of endometrial cancer patients in the study cohort not receiving HRT. Both groups were comparable in terms of parity, tumor grade, depth of myometrial invasion, histology, lymph node status, surgical treatment, concurrent morbidities, and postoperative radiation. The patients receiving HRT were followed for a mean interval of 83 months, and the patients not receiving HRT were followed for a comparable interval of 69 months. Patients using HRT had a longer disease-free interval compared with nonusers of hormones. Among the 150 patients, only 8 patients had stage III disease (115).
In a Committee Opinion by the American College of Obstetricians and Gynecologists Committee of Gynecologic Practice, the members announced that “the decision to use HRT in these women (endometrial cancer patients) should be individualized on the basis of potential benefit and risk to the patient” (117). The Gynecologic Oncology Group (GOG) initiated a multicenter randomized, controlled trial aimed at an enrollment of 2,100 patients with stage I and II endometrial cancer. The study was closed after enrollment of 1,200 patients, because the majority of the patients were low-risk with stage IA and G1 disease. In the absence of adequate enrollment of more intermediate-to-high risk early-stage endometrial cancer patients, the number of recurrences used in the ad hoc sample-size determination would be underestimated, leading to suboptimal power for the clinical trial. With the closure of this GOG protocol, the safety of exogenous estrogen use in women who have undergone surgical management of endometrial cancer may never be fully ascertained.
Oral Contraceptives
Multiple epidemiologic studies have revealed that OCPs are not a risk factor for endometrial cancer, but rather a means of prevention (see below).
Selective Estrogen Receptor Modulators
The antiestrogen tamoxifen has been used in the adjuvant therapy of advanced breast cancer for years, and it has recently been evaluated as a chemopreventive agent in patients at a high risk for developing this malignancy. Tamoxifen increases the risk of second primary malignancies at other sites. A randomized, controlled trial involving 2,729 women performed by the Stockholm Breast Cancer Study Group revealed that patients given tamoxifen (40 mg/day) had a nearly sixfold increased risk of endometrial cancer, and a threefold increased risk of gastrointestinal cancer (118,119). These findings were confirmed in the National Surgical Adjuvant Breast and Bowel Project (NSABP). In this study, 2,843 patients with node-negative invasive breast cancer were randomized to receive tamoxifen (40 mg/day) or placebo. The RR of endometrial cancer in patients receiving tamoxifen was 7.5 (95% CI, 1.7–32.7) (120), compared with placebo (118). When the population-based rates of endometrial cancer from another NSABP trial (B-06), and data from the Surveillance, Epidemiology, and End Results (SEER) program were used in the calculation of risk, the RRs of endometrial cancer associated with tamoxifen use were 2.3 and 2.2, respectively. In addition, the average annual HR during follow-up was only 1.6 out of 1,000 in the group receiving tamoxifen (118,120), suggesting that routine screening would not be cost-effective.
Other selective estrogen receptor modulators (SERMs) are being evaluated as adjuvant treatment for breast cancer in an attempt to avoid the increased risk of secondary malignancies, while maintaining the same or greater effectiveness. Results from the Multiple Outcomes of Raloxifene Evaluation (MORE) study indicate that patients receiving raloxifene had a lower incidence of breast cancer without any increase in endometrial cancer risk (118,121). However, the primary endpoint in this investigation was risk reduction of fracture in postmenopausal women with osteoporosis. In the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 trial, investigators found that raloxifene is as effective as tamoxifen in reducing the risk of invasive breast cancer. Although the overall risk of developing endometrial cancer was similar in patients on raloxifene versus tamoxifen (RR, 0.62; 95% CI, 0.35–1.08), the incidence of hyperplasia was lower in the raloxifene group (RR, 0.16; 95% CI, 0.09–0.29) (122). Several other SERMs are under investigation. The results of ongoing clinical trials will confirm these preliminary findings and establish the applicability of this contemporary generation of SERMs.
Colon
Hormone Replacement Therapy
Multiple epidemiologic studies have revealed that HRT is not a risk factor for colon cancer, but rather protects against this malignancy (see below). The WHI randomized controlled trial revealed that colorectal cancer was reduced among patients who received CEE plus MPA (123). But the interpretation was obscured by a later stage at diagnosis. In the trial involving patients with a history of hysterectomy, patients receiving CEE compared with placebo had similar incidences of colorectal cancer (92). Extended follow-up of the patients from the WHI intervention and post-intervention trials over 8 years revealed that the incidence of colon cancer was still decreased among patients receiving CEE plus MPA compared with placebo (87).
Oral Contraceptives
Multiple epidemiologic studies have revealed that OCPs are not a risk factor for colon cancer, but rather protect against this malignancy (See below).
SERMs
In vitro evidence has revealed that both tamoxifen and raloxifene are effective in reducing cell proliferation and viability of colon cancer cell lines, suggesting a possible application in the prevention of colon cancer (124). Unfortunately, the available epidemiologic evidence is limited and controversial regarding the issue of tamoxifen’s effects on colorectal cancer risk. A meta-analysis by the Stockholm Breast Cancer Study Group analyzed 4,914 postmenopausal women participating in one of three Scandinavian clinical trials evaluating adjuvant tamoxifen: the Stockholm Trial, the Danish Breast Cancer Group Trial, and the South-Swedish Trial (119). The joint analysis of these three trials revealed that adjuvant tamoxifen therapy in breast cancer patients increased the risk of colorectal cancer (RR, 1.9; 95% CI, 1.1–1.3). In contrast, findings from the NSABP B-14 and the SEER program (125) have failed to confirm an increased risk of colorectal cancer among patients receiving adjuvant tamoxifen therapy. Additional epidemiologic trials are needed to resolve the issue of adjuvant tamoxifen therapy and the risk of secondary sporadic colorectal cancers.
Although some epidemiologic evidence suggests that tamoxifen may be associated with an increased risk of sporadic colorectal cancer, SERMs have been used in the treatment of desmoid tumors associated with hereditary colorectal cancer. Colorectal cancer includes 2 main mechanisms of inheritance: familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC). Desmoid tumors are fibroaponeurotic tumors that occur rarely in the general population but can be found in 12% to 17% of patients with FAP (126). Tamoxifen has been shown to have growth inhibition in desmoid tumor cell lines in an E2-independent mechanism (127). According to the practice guidelines of the Standard Task Force of Colon and Rectal Surgeons, high-dose tamoxifen (120 mg/day) and other SERMs may be used in the treatment of aggressive desmoid tumors. This recommendation is weak (level III) and based on limited descriptive European case series (128,129). The use of tamoxifen in the prevention or treatment of primary colorectal cancer is not advocated.
Phytoestrogens
Increased rates of breast, colon, ovarian, and endometrial cancer are found in Western societies. Phyto-E2s are possible dietary mediators of increased cancer risk that are subdivided into two groups: isoflavonoids and lignans. Isoflavonoids are compounds with inherent estrogenic activity that can lead to low IGF expression and inhibition of aromatase and growth factors, resulting in a possible chemoprotective effect in the breast. Lignans are compounds formed from plant lignan precursors by intestinal microflora. Both isoflavonoids and lignans are found in foods such as whole grain rye bread, soybeans, and red clover. Studies suggest that the development period during which the isoflavonoid is ingested is important in modulation of future cancer risk. Ingestion of soy before or during adolescence may decrease the risk of future breast cancer. However, there is no convincing evidence indicating that soy or other isoflavonoids are protective against breast cancer or colon cancer if ingested during adulthood (118,130). There is a paucity of data regarding the association between isoflavonoids and other cancers affecting women. A case-control study using a Hawaiian group revealed that the high consumption of soy products in adults significantly decreased the risk of endometrial cancer, even after accounting for confounding influences (118,131). Large, population-based studies are needed to determine the effects of isoflavonoids on specific cancer risk, particularly since they are becoming a more common component of the American diet.
Lignans can be indirectly measured via urinary enterolactone. Although women with breast cancer have significantly lower levels of urinary enterolactone, it is unknown whether higher levels of enterolactone are protective against breast cancer or whether they are a marker for an unknown chemoprotective compound associated with a healthier diet. High enterolactone production has been associated with inhibition of colon cancer in animal models, but elevated levels have not been confirmed to be protective against colon cancer in humans (118,130). In contrast, obesity as well as increased dietary fat intake is associated with decreased levels of urinary lignans, suggesting that the increased risk of cancer associated with obesity may be in part related to lignan production.
HORMONES AND THE PREVENTION OF HUMAN CANCERS
Chemoprevention can be defined as the use of specific natural or synthetic chemical agents to reverse, suppress, or prevent the progression toward malignancies. Human carcinogenesis proceeds through multiple discernible stages of molecular and cellular alterations, thus providing the scientific rationale for clinical cancer chemoprevention. The necessary requirements for evaluation of chemoprevention include (a) identification of high-risk individuals based on family history and/or genetic mutations; (b) identification of putative surrogate endpoints or biomarkers for cancer; and (c) the possibility that relatively nontoxic agents may decrease the risk of a given malignancy. Prospective clinical trials are mandatory for evaluating potential strategies for preventing human malignancies.
Breast Cancer Chemoprevention
SERMs
Reducing the incidence of breast cancer has the potential to provide a major impact on the morbidity of the disease and its treatment, cost to the individual and to society, and overall cancer mortality. Epidemiologic studies indicate that E2-mediated events play a role in the development of breast cancer. Tamoxifen, a SERM, has been utilized as a chemopreventive agent for breast cancer in four randomized, prospective, placebo-controlled clinical trials. Raloxifene, a second-generation SERM, was investigated in the MORE trial, in which the primary endpoint was risk reduction of fracture in postmenopausal women with osteoporosis; the incidence of breast cancer was a secondary endpoint. These 5 studies are reviewed and summarized below.
National surgical adjuvant breast and bowel project breast cancer prevention trial (P-1). In 1992, the National Cancer Institute, in collaboration with the NSABP, initiated the Breast Cancer Prevention Trial (BCPT, P-1) (132). The primary goal was to determine whether tamoxifen administered for 5 years prevented breast cancer in women at high risk, including women 60 years or older, women 35 to 59 with a 5-year predicted risk of breast cancer of at least 1.66%, or women who had a history of lobular carcinoma in situ (LCIS). Risk was estimated using the Gail model (133). Each of the 13,388 women enrolled was randomly assigned to receive tamoxifen 20 mg/day or placebo. The median follow-up time was 54.6 months, with 175 cases of invasive breast cancer in the placebo group compared with 89 in the tamoxifen group (RR, 0.51; 95% CI, 0.39–0.66; p < 0.00001). Tamoxifen reduced the incidence of ER+ tumors by 69%, but there was no difference in the incidence of ER- tumors. Major toxicities included endometrial cancer (RR = 2.53) and thromboembolic disease (RR = 3.01). The P-1 data has been updated recently, and they have confirmed that the magnitudes of desirable and undesirable effects of tamoxifen are similar to what was observed in the original report (134).
Italian tamoxifen prevention study. Recruitment for this double-blind, placebo-controlled, randomized trial began in October 1992 and ended in July 1997, and included healthy women aged 35 to 70 years (135). The trialist and data-monitoring committee ended recruitment because 26% of women dropped out of this study; 5,408 women were randomized to receive tamoxifen 20 mg/day or placebo for 5 years. Women were allowed to take HRT. The primary endpoints were reduction in the frequency and mortality of breast cancer. At a median follow-up of 46 months, 19 cancers were diagnosed in the tamoxifen arm and 22 among women in the placebo arm (p = 0.6). A 2007 update reported (after 11 years of follow-up) that although there was no overall difference in breast cancer between patients receiving placebo versus tamoxifen, high-risk breast cancers were much less often found among women receiving tamoxifen (RR, 0.24; 95% CI, 0.10–0.59) (136).
Royal Marsden Hospital chemoprevention trial. This trial was initiated in 1986 as a preliminary pilot study for the International Breast Cancer Intervention Study (IBIS-I) (137). The aim of this study was to assess whether tamoxifen would prevent breast cancer in healthy women at increased risk for the disease based on family history only. Each participant had at least one first-degree relative under the age of 50 with breast cancer, one first-degree relative with bilateral breast cancer, or one affected first-degree relative of any age and another affected first-degree or second-degree relative. The women were allowed to take HRT during this study. The participants, 2,494 women between the ages of 30 and 70, were randomized to tamoxifen 20 mg/day or placebo. The median follow-up was 70 months, and 2,471 of the women were analyzed. The frequency of breast cancer was the same for women receiving tamoxifen and placebo (tamoxifen = 34, placebo = 36; RR, 1.06; 95% CI, 0.7–1.7), and there appeared to be no interaction between the use of HRT and the effect of tamoxifen on breast cancer occurrence. An update of this trial reported that contrary to the initial published findings based on 5-year follow-up, there was a clear reduction of invasive estrogen-positive cancers at 10 years (6.2% vs. 5.2%), and the effect was even larger at 15 years (9.6% vs. 7.5%) (138).
International breast cancer intervention study (IBIS). Randomization occurred to tamoxifen 20 mg/day or placebo for 7,152 women, aged 35 to 70 years and at high risk for breast cancer (139). Eligible women had risk factors for breast cancer indicating at least a twofold relative risk among those aged 45 to 70 years, a fourfold relative risk among those aged 40 to 44 years, and a tenfold relative risk among those aged 35 to 39 years. The women enrolled in this study were at moderately increased risk of developing breast cancer, with 60% of the study cohort having a 10-year risk ranging from 5% to 10%. The IBIS-I investigators used a model to predict the absolute 10-year risk of developing breast cancer, but the details of their model have not been published. Use of HRT was permitted, and approximately 40% of women used such therapy at some point during the trial. The primary endpoint was the incidence of breast cancer, including ductal carcinoma in situ (DCIS). At a median follow-up of 50 months (7,139 women analyzed), 69 women in the tamoxifen group compared with 101 women in the placebo group developed breast cancer (overall, 32% reduction in breast cancer rate; 95% CI, 8–50; p = 0.01). Further long-term follow-up has revealed that for ER–positive invasive tumors, the cumulative incidence in the IBIS-I was reduced after 5 years from 2.0% in the placebo arm to 1.5% in the tamoxifen arm, but after 10 years the values were 4.3% versus 2.9%, respectively (140).
Multiple outcome of raloxifene evaluation. The MORE study was a multicenter, randomized, double-blind trial, raloxifene 60 or 120 mg/day or placebo for 3 years, with the primary endpoint of rate of fracture in postmenopausal women with osteoporosis (141). Breast cancer incidence was a secondary endpoint. A total of 7,705 women were enrolled, all of whom were at least 2 years postmenopausal and no older than 80 years of age. Women who were taking E2 during the previous 6 months were excluded, and 12.3% of women reported a family history of breast cancer. There were 27 cases of invasive breast cancer in the placebo group compared with 13 in the raloxifene group (RR 0.24; 95% CI, 0.13–0.44; p < 0.001). There was no difference in breast cancer incidence between the 2 doses of raloxifene. Raloxifene reduced the risk of invasive ER+ breast cancer by 90% (RR 0.10; 95% CI, 0.04–0.24) but did not reduce the risk of ER- breast cancer. The 4-year results from this study, with extension to 4 years after initiation of the trial, were consistent with the results based on the 3-year follow-up (142). The Continuing Outcomes Relevant to Evista (CORE) trial examined the effect of an additional 4 years of raloxifene therapy on the incidence of breast cancer in women in MORE. Over the 8 years of both trials, the incidence of invasive breast cancer and ER–positive breast cancer were significantly reduced, but the incidence of ER-negative tumors was not (143).
The National Surgical Adjuvant Breast and Bowel Project (NSABP) protocol P-2 or the Study of Tamoxifen and Raloxifene (STAR) Trial. This study, opened to accrual on July 1, 1999, was powered to demonstrate superior efficacy or equivalence of either tamoxifen (20 mg/day) or raloxifene (60 mg/day) in reducing the incidence of primary breast cancer. The trial enrolled 19,747 women, and data are available for 19,471 of them—9,726 women in the tamoxifen arm and 9,745 women in the raloxifene arm. All women had a projected 5-year risk of developing breast cancer of 1.66% or higher as determined by the Gail model. Eligible women included postmenopausal women 35 years or older with no prior history of invasive breast cancer or DCIS. Postmenopausal women aged 35 or older with a history of LCIS were also eligible. Preliminary results of the STAR trial revealed that tamoxifen and raloxifene were as effective as tamoxifen in reducing the risk of invasive breast cancer (RR, 0.70; 95% CI) (122). Recent updates of the STAR trial have shown after 8 years of follow-up that raloxifene is less effective than tamoxifen in reducing invasive cancer, but it is less often associated with endometrial cancer or endometrial hyperplasia (144). In addition, patients treated with raloxifene are diagnosed with endometrial polyps and have fewer hysterectomies while in treatment, compared with women who receive tamoxifen (145).
Aromatase Inhibitors
Aromatase inhibitors (AIs) are being considered for use in breast cancer chemoprevention. In contrast to SERMs, which are competitive inhibitors of E2 at its receptor, AIs suppress plasma E2 levels by inhibiting or inactivating aromatase, the enzyme responsible for the synthesis of E2s from androgenic substrates. This class of compounds has effectively challenged tamoxifen for use as adjuvant therapy in postmenopausal women with ER+ breast cancers, who constitute the majority of patients with breast cancer. Evaluating women with early-stage breast cancer, the ATAC (Arimidex, Tamoxifen, Alone or in Combination) Trialists’ Group found that the incidence of contralateral invasive breast cancer was lower in those receiving the third-generation AI (anastrozole [Arimidex]), when compared with tamoxifen, after a median of 100 months of follow-up. For estrogen-positive tumors, disease-free survival and time to survival were both improved significantly among patients receiving anastrozole compared with those given tamoxifen (HR, 0.85, 95% CI, 0.76–0.94; and HR, 0.76, 95% CI, 0.67–0.87). Fracture rates were higher among patients receiving anastrozole than those receiving tamoxifen during active treatment, but there was no difference after the treatment was completed (146).
Prophylactic Oophorectomy
The potential role for oophorectomy as chemoprevention (decreased E2 levels) for breast cancer in premenopausal women is an area of active investigation. In genetically uncharacterized premenopausal women, oophorectomy is associated with a 50% reduction in breast cancer incidence, but it also incurs the associated negative effects of a surgically induced early menopause (147,148). In this patient population, where the overall incidence of breast cancer is 1 in 8, it is not standard practice to offer oophorectomy for breast cancer prevention. However, in premenopausal women with germ-line BRCA1 and BRCA2 mutations, the cumulative lifetime risk (to 70 years of age) of invasive breast cancer is 60% to 85%, allowing for more serious consideration of prophylactic oophorectomy as a chemoprevention. The prevalence of germ-line BRCA1 or BRCA2 in the general population is 0.1% to 0.2%, contributing to a small fraction of all cases of breast cancer, but as many as 10% of cases diagnosed in women younger than 40 years of age and approximately 75% of familial cases. Two publications support the practice of recommending prophylactic oophorectomy after the completion of childbearing in premenopausal women (149,150). Kauff et al. in a prospective study, reported on 170 carriers of BRCA mutations with a mean follow-up of 24.2 months, 98 of whom underwent prophylactic salpingo-oophorectomy (149). A 70% risk reduction in breast cancer incidence was identified. Rebbeck et al. in a multicenter retrospective analysis, reported on 551 carriers of BRCA mutations with 11 years of follow-up, 259 of whom underwent a prophylactic salpingo-oophorectomy. A 53% risk reduction in breast cancer incidence was identified (150). The reduced risk of a breast cancer diagnosis after salpingo-oophorectomy in women with BRCA mutations confirms an earlier report of a 47% reduction in this patient population (151). The age at risk-reducing depends on the type of mutation and the family history (152
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


