Chapter 501 Histiocytosis Syndromes of Childhood
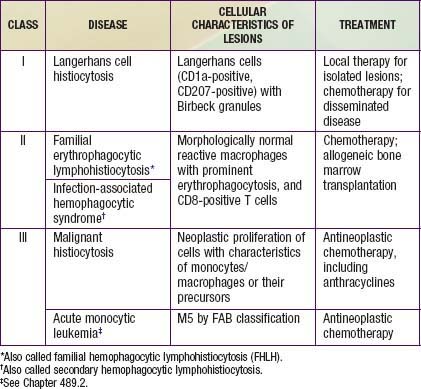
The childhood histiocytoses constitute a diverse group of disorders, which, although individually rare, may be severe in their clinical expression. These disorders are grouped together because they have in common a prominent proliferation or accumulation of cells of the monocyte-macrophage system of bone marrow origin. Although these disorders sometimes are difficult to distinguish clinically, accurate diagnosis is essential nevertheless for facilitating progress in treatment. A systematic classification of the childhood histiocytoses is based on histopathologic findings (Table 501-1). A thorough, comprehensive evaluation of a biopsy specimen obtained at the time of diagnosis is essential. This evaluation includes studies such as electron microscopy and immunsotaining that may require special sample processing.
Classification and Pathology
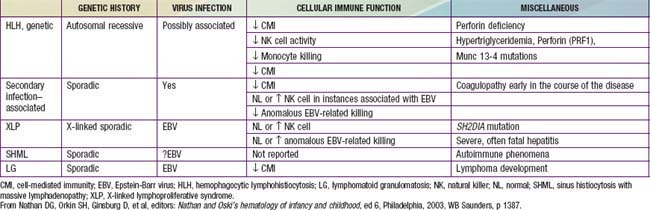
In contrast to the prominence of an antigen-presenting cell (the Langerhans cell) in the class I histiocytoses, the class II histiocytoses are nonmalignant proliferative disorders that are characterized by accumulation of antigen-processing cells (macrophages). Hemophagocytic lymphohistiocytoses (HLH) are the result of uncontrolled hemophagocytosis and uncontrolled activation (upregulation) of inflammatory cytokines similar to the macrophage activation syndrome (see Table 149-5). Tissue infiltration by activated CD8 T lymphocytes, activated macrophages, and hypercytokinemia are classic features. With the characteristic morphology of normal macrophages by light microscopy, these phagocytic cells are negative for the markers (Birbeck granules, CD1a-positivity, CD207-positivity) characteristic of the cells found in LCH. The 2 major diseases among the class II histiocytoses have indistinguishable pathologic findings. One is familial hemophagocytic lymphohistiocytosis (FHLH), previously called familial erythrophagocytic lymphohistiocytosis (FEL), which is the only inherited form of histiocytosis and is autosomal recessive. Some specific genes involved with FEL include mutations of perforin, Munc 13-4, and Syntaxin-11 and all are related to pathways of granule-mediated cellular cytotoxicity. The other is the infection-associated hemophagocytic syndrome (IAHS), also called secondary hemophagocytic lymphohistiocytosis (Table 501-2). Both diseases are characterized by disseminated lesions that involve many organ systems. The lesions are characterized by infiltration of the involved organ with activated phagocytic macrophages and lymphocytes, in which the lymphocyte (cytolytic pathway) defects are considered to be the primary abnormality. These diseases are grouped together under the term hemophagocytic lymphohistiocytosis (HLH) (Table 501-3).
Table 501-2 INFECTIONS ASSOCIATED WITH HEMOPHAGOCYTIC SYNDROME
VIRAL
BACTERIAL
FUNGAL
MYCOBACTERIAL
RICKETTSIAL
PARASITIC
From Nathan DG, Orkin SH, Ginsburg D, et al, editors: Nathan and Oski’s hematology of infancy and childhood, ed 6, Philadelphia, 2003, WB Saunders, p 1381.
The class III histiocytoses, in contrast, are unequivocal malignancies of cells of monocyte-macrophage lineage. By this definition, acute monocytic leukemia and true malignant histiocytosis are included among the class III histiocytoses (Chapter 489). The existence of neoplasms of Langerhans cells is controversial. Some cases of LCH demonstrate clonality.
501.1 Class I Histiocytoses
Clinical Manifestations
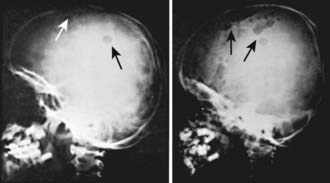
LCH has an extremely variable presentation. The skeleton is involved in 80% of patients and may be the only affected site, especially in children >5 yr of age. Bone lesions may be single or multiple and are seen most commonly in the skull (Fig. 501-1). Other sites include the pelvis, femur, vertebra, maxilla, and mandible. They may be asymptomatic or associated with pain and local swelling. Involvement of the spine may result in collapse of the vertebral body, which can be seen radiographically, and may cause secondary compression of the spinal cord. In flat and long bones, osteolytic lesions with sharp borders occur and no evidence exists of reactive new bone formation until the lesions begin to heal. Lesions that involve weight-bearing long bones may result in pathologic fractures. Chronically draining, infected ears are commonly associated with destruction in the mastoid area. Bone destruction in the mandible and maxilla may result in teeth that, on radiographs, appear to be free floating. With response to therapy, healing may be complete.