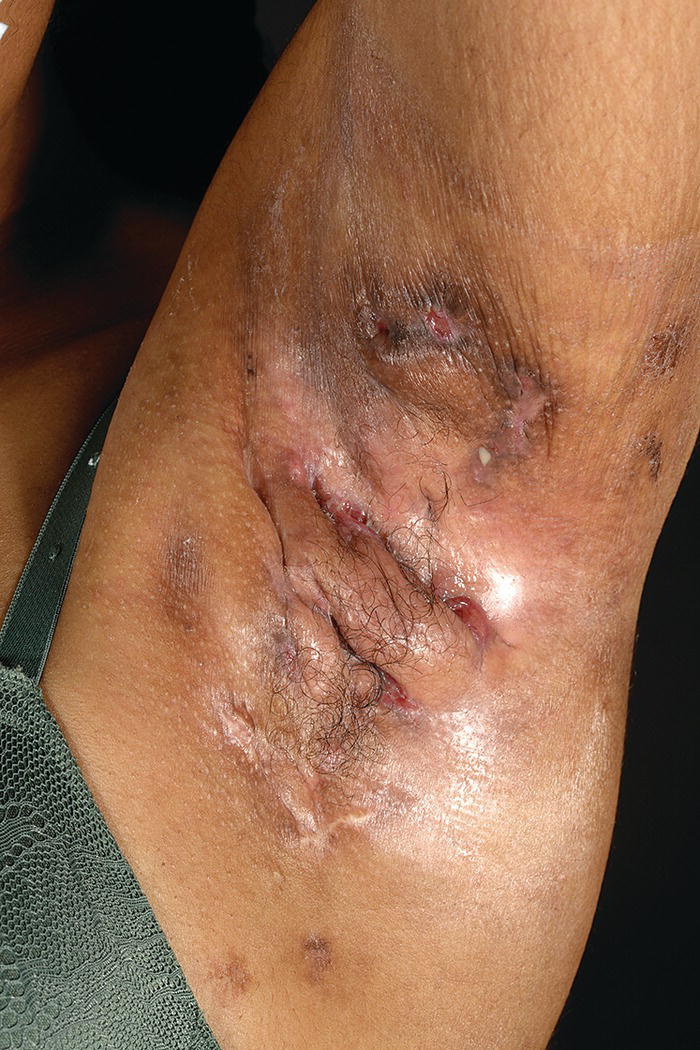
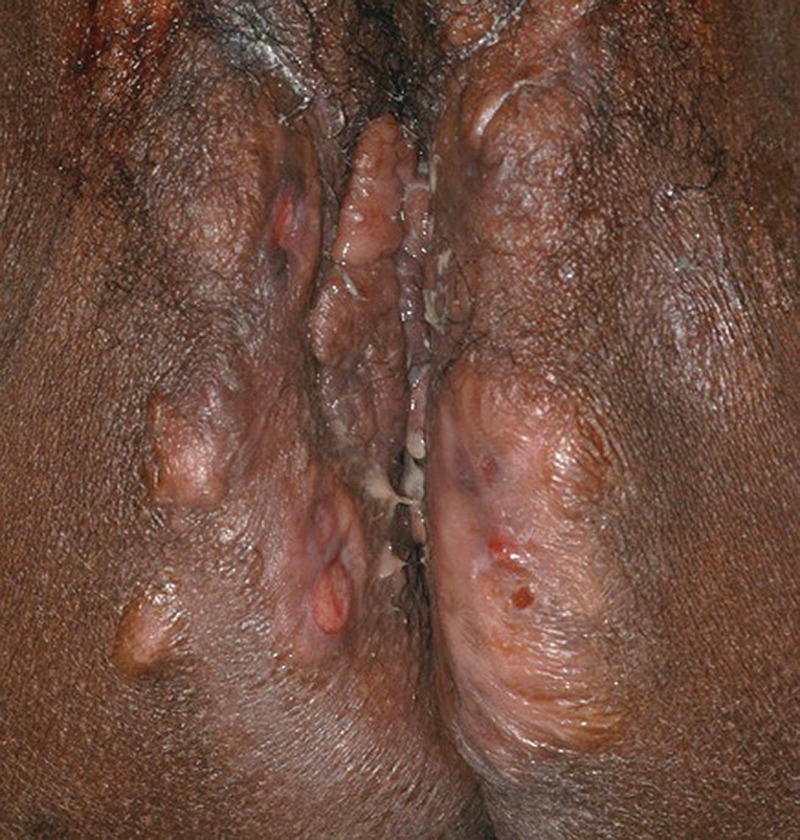
Ellie Rashidghamat Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterised by painful, deep‐seated, inflamed lesions in the apocrine gland‐bearing areas of the body. These lesions include inflammatory nodules, abscesses, sinus tract formation, scarring, and fistulae that show a predilection for intertriginous sites, usually presenting after puberty, most commonly in the axillary, inguinal, and anogenital regions. Sequelae include significant pain, scarring, and psychological distress. HS develops at an average age of 22.1 years. The onset is usually post‐pubertal, but it can occur at any age, and persists for an average of 18.8 years [1]. The 1‐year prevalence is estimated to be 1–4% in European populations [2, 3]and 0.03–0.053% in the United States [4, 5]. Various methodologies are used to estimate the prevalence, and so as a result, there is a wide range. In some studies, there was a reliance on patient self‐reporting, and the method of data collection varied, including surveys and patient health insurance claims, thus introducing variation. Patients first present to a range of medical and surgical specialities (accident and emergency, primary care, genitourinary medicine, gynaecology, dermatology, general, plastic, breast surgery, and urology), which can result in significant diagnostic delay. In addition, low rates of presentation and diagnosis due to factors such as stigma associated with the body sites affected leading to embarrassment, and a lack of clinical knowledge about the condition are further confounding factors. There is a female predominance, with reported female to male ratios as high as 5:1 [6]. There is an inherited component in the aetiology of HS with several mutations reported in the gamma‐secretase genes (PSENEN, PSEN1, and NCSTN) [7–9]. Exactly how those genes result in disease mutations is likely to be via the gamma‐secretase‐NOTCH signalling pathway. The gamma‐secretase complex (GSC) is involved in NOTCH signalling, which has been shown to be heavily involved in the maintenance of the outer and inner root sheath of hair follicles [10]. Mutations have been identified in the PSTPIP1 gene in cases of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) and pyogenic arthritis, pyoderma gangrenosum, acne, and suppurative hidradenitis (PAPASH) syndromes [11]. However, the majority of HS patients do not have identifiable mutations. A family history of HS is reported in approximately a third of patients [12]. The aetio‐pathogenesis of HS is multifactorial and incompletely understood. It was initially thought to be a disease of the apocrine gland [13], due to the presence of a peri‐glandular inflammatory infiltrate in affected skin and involvement of apocrine‐gland‐bearing sites [14, 15]. Histopathological evidence, however, has implicated the hair follicle rather than the apocrine gland in HS pathogenesis, suggesting that follicular plugging is the primary histopathological mechanism underlying HS [16, 17], with a retrospective pathological study identifying follicular occlusion in all stages of disease [17]. Subsequent to follicular occlusion and dilatation, other endogenous factors, which are likely to include genetic disposition, enhance the risk of infundibular keratinisation and cyst formation. In addition, exogenous factors including smoking and obesity are important. Occlusion of the hair follicle leads to dilatation and rupture [18]. This rupture results in a secondary dispersion of the follicular contents including keratin fibres and commensal flora into the dermis, and damage‐associated molecular patterns (PAMPs/DAMPs) trigger a significant immune response with activation of multiple inflammatory pathways, particularly Th17/IL‐23, inflammasomes, and innate toll‐like receptors such as TLR2 [18]. The resultant involvement of monocytes, neutrophils, multinucleated giant cells, B‐cells, plasma cells, T‐cells, and natural killer cells leads to the clinical signs. These include erythematous nodules and abscess formation with chronic inflammation developing into sinus tracts/tunnel formation [18]. Continuous activation of the immune system with an imbalance of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinase (TIMPs) together with an increase in the activity of pro‐fibrotic factors such as tissue growth factor (TGF)‐ß 1‐2‐3 leads to scarring and fistulae formation. Biofilm‐producing bacteria thrive in this environment, triggering further inflammation with pus and discharge [18]. Elevated levels of pro‐inflammatory cytokines, including tumour necrosis factor (TNF)‐α, interleukin (IL)‐1ß, IL‐17 and interferon (IFN)‐γ, have been observed in HS lesions [19] in addition to T helper (Th) cell accumulation [20]. There is also a significant association between C‐reactive protein (CRP) levels and neutrophil count with HS disease severity [21]. The main histological changes include folliculitis and ductal occlusion. There is secondary involvement of the apocrine and eccrine glands [22]. Sinus tracts lined by keratinised epithelium are formed (Figure 25.1) and are seen more often if there is evidence of poral occlusion of the duct or epithelial cysts. A dense inflammatory infiltrate is common (Figure 25.2). There is often distension of apocrine glands and accumulations of polymorphs, which tend to migrate to adjacent tissue. The gland may become necrotic, with an infiltrate of lymphocytes, plasma cells, and macrophages. Ultimately, the changes lead to abscesses, fibrosis, foreign body reactions, and sometimes pseudoepitheliomatous hyperplasia [22]. Most cases of HS occur after puberty, with pre‐menstrual flares being common in women [23] and tends to involute post menopause, although some cases do continue into the post‐menopausal period [24]. There is also an association between HS and polycystic ovarian syndrome [25]. A significant subset of women report amelioration of their HS symptoms during pregnancy [26]. There are differing clinical presentations between the sexes. In women, the anterior aspects of the body are affected including the groin, thighs, and breasts; however, in males, the gluteal areas including the buttocks, perineal, and perianal areas are more commonly involved. Axillary involvement is equally distributed between men and women [27]. Men overall have more acne, pilonidal cysts, and involvement of atypical sites such as post‐auricular. However, women are more often affected with concomitant acne. Figure 25.1 Hidradenitis suppurativa. Marked inflammation, including suppuration, beneath and adjacent to a sinus tract, with surrounding fibrosis. Figure 25.2 Hidradenitis suppurativa. Dense inflammatory infiltrate containing neutrophils, plasma cells, and giant cells; the latter often reflects previous follicular rupture. The clinical hallmarks of the disease are recurrent painful inflammatory nodules, abscesses, sinus tracts, and scarring. Affected sites usually involve flexural areas including the axillae (Figure 25.3), groins, perineum, buttocks, medial thighs, submammary region, abdominal fold, and posterior auricular skin. An important characteristic and diagnostic feature is the presence of bridged scars and comedonal plugs. Inflammatory nodules are often the first clinical sign and can last for several days to several months. Inflammatory lesions can be fixed or migratory. Following on from this, these nodules can progress to form abscesses that then yield a purulent discharge (Figure 25.4). Sinus tracts or draining tunnels can persist for months to years. Development of multiple inflammatory nodules and abscesses in an anatomical location can give rise to the formation of draining tunnels. These tunnels drain purulent and serosanguinous discharge. Comedones often denote damage to the folliculopilosebaceous unit [28]. Closed comedones result from keratin production from the follicular epithelium above the damaged sebofollicular junction. Scarring can take on different morphologies ranging from pitted scars, to fibrotic bands and rope like scarring and indurated scarred plaques. (Figure 25.5). The diagnostic criteria in HS follows a thorough patient history and observation of clinical signs. Three features that support a diagnosis of HS include typical lesions (as described above), typical sites, and chronic lesions (usually longer than 6 months, or more than five inflammatory lesions over lifetime). The Hurley staging system is used to score clinical severity in HS (see Table 25.1). Most patients are Hurley stage 1, with a small minority being Hurley stage 3 [27]. Figure 25.3 Hidradenitis suppurativa: axillary involvement with abscesses and sinus tracts. Figure 25.4 Nodules, abscesses, and draining sinuses. Other important patient assessment measures include lesion count, pain scoring (such as the use of a visual analogue scale), and the Dermatology Life Quality Index (DLQI). The Hidradenitis Suppurativa Clinical Response (HiSCR) was developed retrospectively from a randomised controlled trial and is a binary measure to assess treatment response. It calculates the number of abscesses and inflammatory nodules, designated ‘ANs’, in addition to the number of draining sinus tracts present in a patient with HS. One achieves HiSCR if there is at least a 50% reduction in ANs, with no increase in the number of abscesses or draining sinuses, relative to the baseline [29]. Figure 25.5 Thickened scarring on mons pubis in hidradenitis suppurativa. Table 25.1 Hurley staging system in hidradenitis suppurativa. The differential diagnosis for HS can include a primary cutaneous infection such as folliculitis, furuncles, or carbuncles arising from hair follicles. Acne has several overlapping features with HS including comedones, inflammatory nodules, and scarring; however, acne often affects the face, chest, and back, and HS usually affects intertriginous sites. Pilonidal disease is a sinus tract that arises in the natal cleft, and alongside HS, acne conglobata, and dissecting cellulitis of the scalp, makes up the follicular occlusion tetrad [30]. Crohn’s disease can have overlapping features with HS with abscesses, sinus tracts, and fistulating disease. Cutaneous Crohn’s disease can occur in association with HS, and features classic “knife cut” fissures, undermined ulceration, and oedema of the inguinal and interlabial folds (see Chapter 32).
25
Hidradenitis Suppurativa
Epidemiology
Genetics
Pathophysiology
Histological features
Clinical features


Staging systems

Hurley stage
Clinical features
1
Isolated lesions with no sinus tract formation and absent or minimal scarring
2
Recurrent lesions separated by normal skin with sinus tract formation and scarring
3
Multiple lesions coalescing into inflammatory plaques involving most of the affected region
Differential diagnosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


