Chapter 605 Hereditary Motor-Sensory Neuropathies
605.1 Peroneal Muscular Atrophy (Charcot-Marie-Tooth Disease; HMSN Type I)
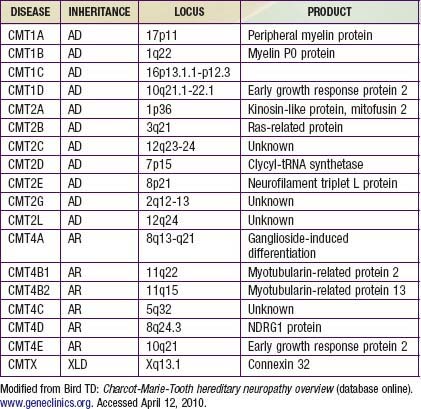
Charcot-Marie-Tooth disease is the most common genetically determined neuropathy and has an overall prevalence of 3.8/100,000. It is transmitted as an autosomal dominant trait with 83% expressivity; the 17p11.2 locus is the site of the abnormal gene. Autosomal recessive transmission also is described but is rarer. The gene product is peripheral myelin protein 22 (PMP22). A much rarer X-linked HMSN type I results from a defect at the Xq13.l locus, causing mutations in the gap junction protein connexin-32. Other forms have been reported (Table 605-1).
Clinical Manifestations
Davidenkow syndrome is a variant of HMSN type I with a scapuloperoneal distribution.



