Chapter 469 Hemostasis
The Process
After vascular injury, vasoconstriction occurs and flowing blood comes in contact with the subendothelial matrix (Fig. 469-1). In flowing blood, when exposed to subendothelial matrix proteins, von Willebrand factor (VWF) changes conformation and provides the glue to which the platelet VWF receptor, the glycoprotein Ib complex, binds, tethering platelets to sites of injury. When the VWF receptor binds its ligand, complex signaling occurs from the outside membrane receptor to intracellular pathways, activating the platelets and triggering secretion of storage granules containing adenosine diphosphate (ADP), serotonin, and stored plasma and platelet membrane proteins. Upon activation, the platelet receptor for fibrinogen, α2bβ3, is switched on (“inside out” signaling) to bind fibrinogen and triggers the aggregation and recruitment of other platelets to form the platelet plug. Multiple physiologic agonists can trigger platelet activation and aggregation, including ADP, collagen, thrombin, and arachidonic acid. Aggregation involves the interaction of specific receptors on the platelet surface with plasma hemostatic proteins, primarily fibrinogen.
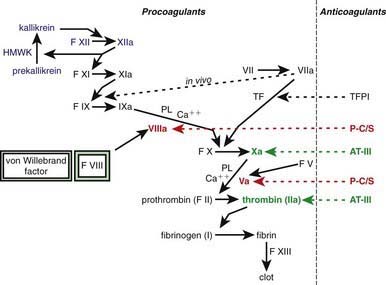
One of the subendothelial matrix proteins that are exposed after vascular injury is tissue factor. Just as exposed subendothelial matrix proteins bind VWF, exposed tissue factor binds to factor VII and activates the clotting cascade, as shown in Figure 469-2. The activated clotting factor then initiates the activation of the next sequential clotting factor in a systematic manner. Our understanding of the sequence of steps in the cascade followed assignment of the numerals for the clotting factors for the participant proteins, and thus the sequence seems “out of numerical order.” During the process of platelet activation, internalized platelet phospholipids (primarily phosphatidylserine) become externalized and interact at 2 specific, rate-limiting steps in the clotting process—those involving the cofactors factor VIII (X-ase complex) and factor V (prothrombinase complex). Both of these reactions are localized to the platelet surface and bring together the active enzyme, an activated cofactor, and the zymogen that will form the next active enzyme in the cascade. This sequence results in amplification of the process, which supplies a burst of clotting where it is physiologically needed. In vivo, autocatalysis of factor VII generates small amounts of VIIa continuously, so the system is always poised to act. Near the bottom of the cascade, the multipotent enzyme thrombin is formed. Thrombin converts fibrinogen into fibrin, activates factors V, VIII, and XI, and aggregates platelets. Activation of factor XI by thrombin amplifies further thrombin generation and contributes to inhibition of fibrinolysis. Thrombin also activates factor XIII. The stable fibrin-platelet plug is ultimately formed through clot retraction and cross linking of the fibrin clot by factor XIIIa.
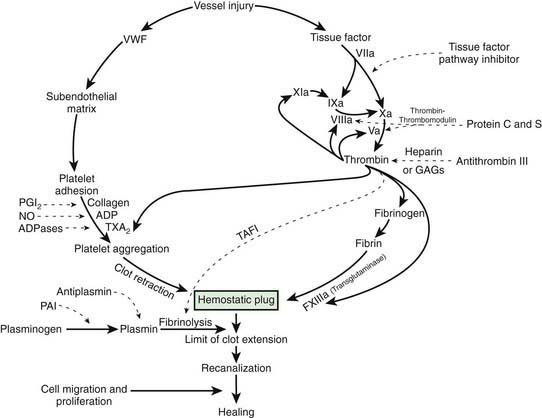
Virtually all procoagulant proteins are balanced by an anticoagulant protein that regulates or inhibits procoagulant function. Four clinically important, naturally occurring anticoagulants regulate the extension of the clotting process. They are antithrombin III (AT-III), protein C, protein S, and tissue factor pathway inhibitor (TFPI). AT-III is a serine protease inhibitor that regulates factor Xa and thrombin primarily and factors IXa, XIa, and XIIa to a lesser extent. When thrombin in flowing blood encounters intact endothelium, thrombin binds to thrombomodulin, its endothelial receptor. The thrombin-thrombomodulin complex then converts protein C into activated protein C. In the presence of the cofactor protein S, activated protein C proteolyses and inactivates factor Va and factor VIIIa. Inactivated factor Va is, in fact, a functional anticoagulant that inhibits clotting. TFPI limits activation of factor X by factor VIIa and tissue factor and shifts the activation site of tissue factor and factor VIIa to that of factor IX (see Figs. 469-1 and 469-2).



