11 Hematology and Oncology
Hematology
Red Blood Cells
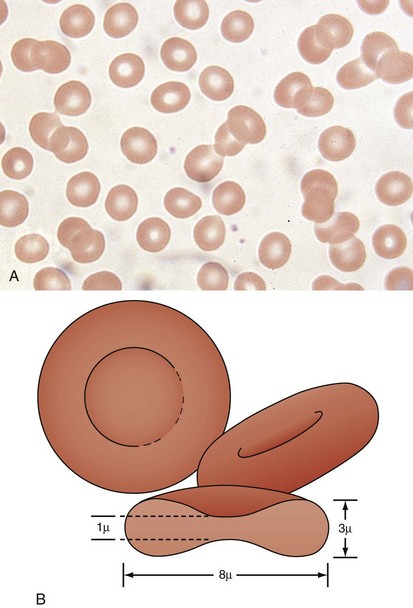
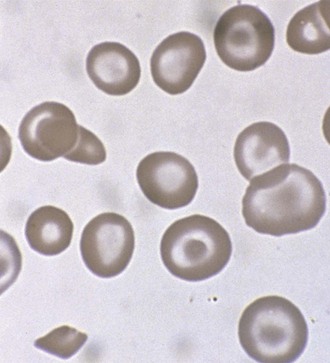
A peripheral smear is made with a small droplet of blood on a glass slide. A second slide can be used to smear the blood in a manner that covers one half to two thirds the length of the slide. A well-made smear should not have lines or holes, nor should it reach either end of the slide completely. Commonly, Wright stain is applied after drying to give the cells their characteristic appearance (Fig. 11-1). In general, a normal red blood cell has approximately the same size as a lymphocyte nucleus. Red cells all should be the same relative shape and size, with an area of central pallor that is approximately one third the diameter of the cell (Fig. 11-2). A variety of terms are available to describe the physical properties of mature red blood cells. Polychromasia refers to variability in cell color, with hypochromia specifically indicating an increased area of central pallor. Anisocytosis indicates variability in cell size. Microcytosis describes a small cell size, whereas macrocytosis refers to large cells. Poikilocytosis represents variability in cell shape. The overall number of red blood cells in the circulation cannot be described reliably by the peripheral smear. Laboratory values can also aid in assessing red blood cell morphology. Two of these have wide usage. The mean corpuscular volume (MCV) is a laboratory measure of cell size, and red cell distribution width (RDW) estimates this difference.
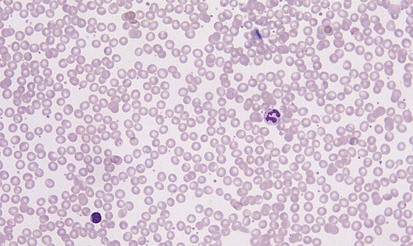
Anemia
Classifications and Symptoms
Increased RBC loss or decreased RBC production can result in an overall decrease in RBC mass below a critical level, leading to anemia. This is the most common abnormality of RBCs, and it is identified as a decreased hemoglobin level. At any given age, anemia is defined practically as a value greater than 2 standard deviations below the mean (Table 11-1). However, physiologically, anemia is best defined as a hemoglobin level that is too low to meet tissue oxygen demands. Conceptually, anemia occurs as a result of one of the following: decreased bone marrow production of red blood cells (RBCs) and reticulocytes (Fig. 11-3), increased destruction of mature RBCs peripherally or as precursors in the bone marrow, or hemorrhage. In addition, a combination of these factors can occur.
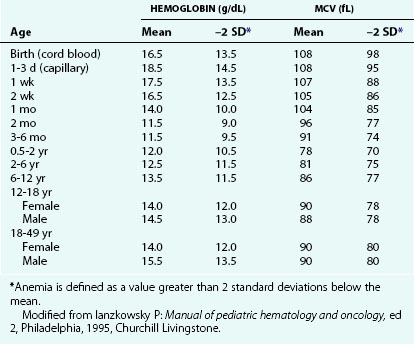
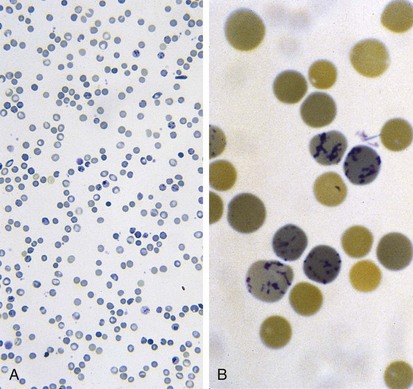
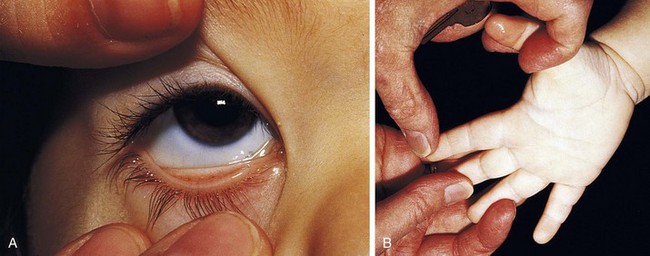
Recognition that a child’s hemoglobin level is abnormal requires knowledge of age, gender, and race-related normal values. Severe anemia from any cause may elicit symptoms of fatigue; decreased appetite; headache; and, in extreme cases, shock, congestive heart failure, or even stroke. Physical examination of the anemic child may reveal pallor, although in the fair-skinned or dark-skinned child this may be easily missed, unless palmar creases or conjunctivae are also examined thoroughly (Fig. 11-4). Vital signs may be normal, but with severe anemia, tachycardia may be present. In practice, it is common for anemias to be classified on the basis of their cellular morphology, especially size and coloration. The most common scenario encountered in pediatrics is a microcytic anemia due to iron deficiency.
Iron Deficiency
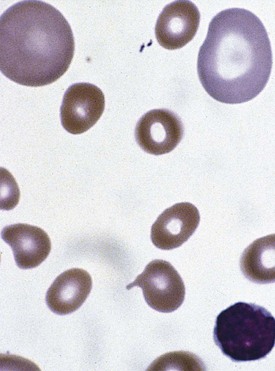
A lack of iron leads to failure of hemoglobin production. Affected children may manifest a peculiar physical finding termed koilonychia, or “spooning” of fingernails and toenails (Fig. 11-5). Glossitis may be observed in iron deficiency and other nutritional deficiencies. Iron deficiency, however, is only a laboratory finding, not a diagnosis. The causes of iron deficiency must be elucidated through a careful history and physical examination. Although poor nutrition is the most common cause, other causes such as hemorrhage or malabsorption must also be considered. Iron deficiency anemia occurs in infants whose rapidly increasing RBC mass outstrips the dietary iron intake. Because the normal full-term infant has adequate iron reserve to accommodate the increasing RBC mass through the first 5 months of life, iron deficiency is usually not seen until the second half of the first year of life. It is detected most often in the 10- to 18-month-old child who ingests large volumes of cow’s milk and little else. Whole cow’s milk not only is deficient in dietary iron but often leads to an enteropathic condition with occult gastrointestinal blood loss, exacerbating the child’s iron-deficient status. A second peak of iron deficiency anemia is seen during adolescence in girls.
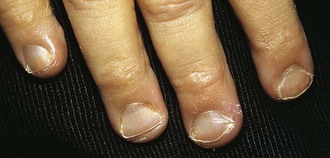
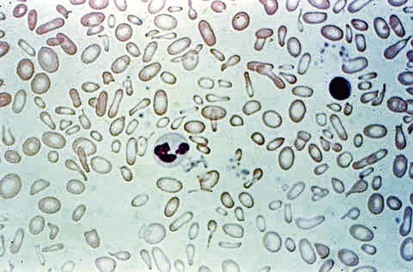
Figure 11-6 shows a peripheral blood smear of a child with iron deficiency anemia. The RBCs are microcytic and hypochromic. The number of platelets often is increased (particularly when an enteropathic condition is present), although it may be normal or even decreased. In severe iron deficiency anemia, anisocytosis and poikilocytosis may be prominent. Nonspecific abnormalities of RBC morphology, such as the presence of micro-ovalocytes (see Fig. 11-6) or basophilic stippling (see Fig. 11-8), may also occur. Bizarre RBC shapes are due to a three-dimensional change in structure when viewed in a two-dimensional plane of the light microscope. When viewed in two dimensions, the cells appear ovoid.
Lead Poisoning
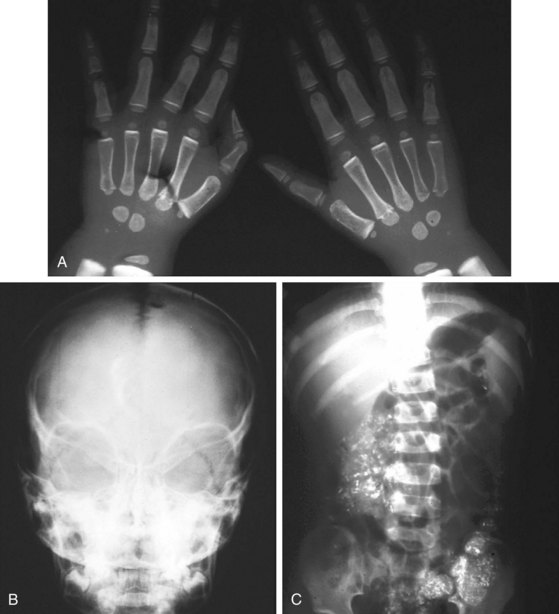
Lead intoxication also leads to microcytic anemia. However, nonhematologic manifestations of lead intoxication, particularly neurologic complications, often dominate the picture. The spectrum of clinical presentations of lead intoxication ranges from vague symptoms of abdominal pain, vomiting, malaise, and behavioral changes to acute encephalopathic conditions, with rapid progression to coma and death. Late physical findings may include papilledema. Significant radiographic changes are also seen in lead intoxication (Fig. 11-7).
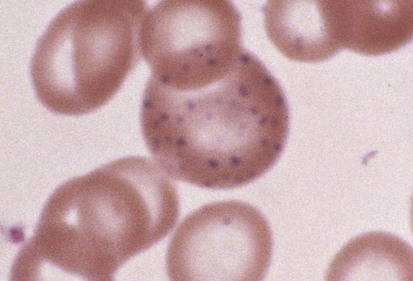
Hematologic abnormalities of lead intoxication are a direct result of the effect of lead on several cellular enzymes involved in heme production. Lead inhibits these enzyme systems, impairing iron use and globin synthesis. Thus despite normal intracellular levels, iron is unable to be incorporated into heme and hemoglobin production fails. Reduced hemoglobin production leads directly to hypochromic, microcytic anemia. Basophilic stippling (Fig. 11-8) is a secondary and inconsistent hematologic manifestation of lead intoxication that occurs as a result of inhibition of yet another RBC enzyme, pyrimidine 5′-nucleotidase. Although basophilic stippling is more prominent in lead intoxication, it is also present in thalassemia and treated iron deficiency. Its presence on the peripheral smear is nonspecific.
Megaloblastic Anemia
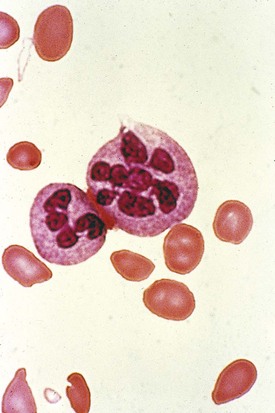
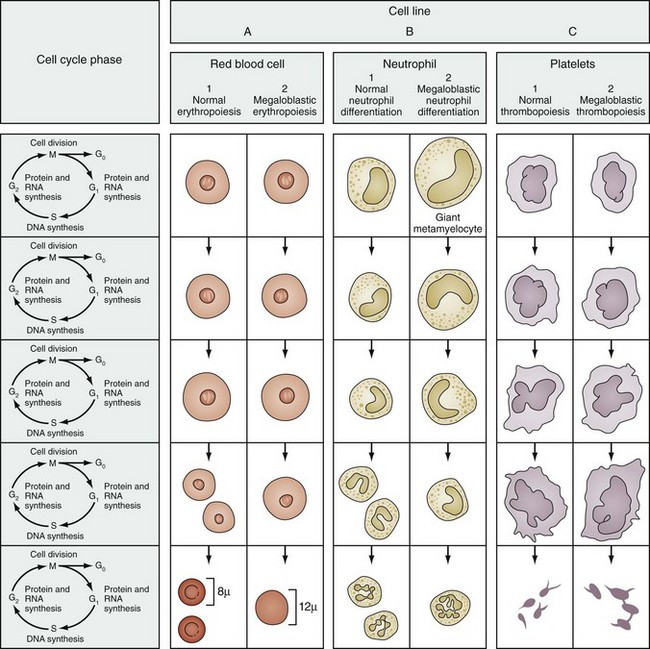
On the peripheral blood smear, the RBCs are large in size (macrocytes) and display a great deal of variation in their shapes (Fig. 11-9). These macrocytic cells are generally normochromic. In addition, all of the actively dividing marrow cells may become involved in the pathologic process. Neutrophils are the second most likely cells to display morphologic abnormalities. These cells become large and show pathognomonic hypersegmentation of the nucleus (Fig. 11-10). Neutropenia is common. The more severe and prolonged megaloblastic anemias ultimately may lead to moderate thrombocytopenia, with large bizarre platelets on the peripheral blood smear. Megaloblastic changes in each cell line are shown in Figure 11-11.
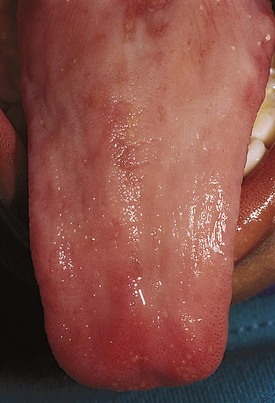
Laboratory diagnosis requires the measurement of folate and vitamin B12 levels in the serum and RBCs. Because a typical Western diet is unlikely to lead to folate or vitamin B12 deficiency, a low level of either should raise questions of altered bioavailability or peculiar diet. Goat milk is folate deficient (although in more recent years many canned goat milk products are folate supplemented), and an infant on a goat milk diet may become folate depleted over time. Fad diets often are not structured thoughtfully and may lead to folate deficiency. Various drugs that decrease folate absorption (phenytoin) or interfere with folate metabolism (methotrexate) may also lead to megaloblastic changes. A pathologic condition of the gastrointestinal tract may cause malabsorption of folate or vitamin B12 and secondary megaloblastic changes. This condition may be iatrogenic if a portion of the intestines is removed for medical reasons. Pernicious anemia is a specific cause due to a deficiency of intrinsic factor, which is required for vitamin B12 absorption. Glossitis (Fig. 11-12) or angular stomatitis, commonly seen in vitamin B12 deficiency, can be helpful physical findings in the diagnostic process.
Pure Red Cell Aplasia
Transient erythroblastopenia of childhood (TEC) occurs in 1- to 4-year-old children and appears 2 weeks to 2 months after a respiratory or gastrointestinal illness. TEC is transient and self-limited, whereas the other forms of red cell aplasia may be chronic. It has been noted that the RBCs in pure red cell aplasia have fetal characteristics, whereas the RBCs of TEC have age-appropriate characteristics (Table 11-2). The cause of TEC remains obscure. TEC may occur after many different viral infections and most likely represents an altered immunity from the infection that affects erythropoiesis. Treatment is supportive and determined by the degree of symptoms.
Table 11-2 Features Differentiating Pure Red Cell Anemia from Transient Erythroblastopenia of Childhood
| disease | ||
|---|---|---|
| RBC Characteristic | Pure Red Cell Anemia | TEC |
| Hemoglobin | Increased fetal | Normal fetal |
| Cellular antigen | i | I |
| Mean corpuscular volume | Increased | Normal |
| RBC enzyme activity | Normal or high | Low |
RBC, red blood cell; TEC, transient erythroblastopenia of childhood.
Hemolytic Anemias
Hemolytic anemias are defined by the premature destruction of red blood cells. This can occur by mechanical, infectious, enzymatic, or immune-mediated mechanisms. Symptoms and severity vary not only among individuals but also between diagnoses. Jaundice, especially scleral icterus, as well as splenomegaly and dark urine can be common presentations during hemolytic episodes. Although all hemolytic anemias may have acute exacerbations, some also feature more chronic phases of red cell breakdown. The history of individual patients, laboratory studies, and blood cell morphology are important factors in making the accurate diagnosis. Treatments can vary; often, splenectomy may be a consideration. However, splenectomy in children is fraught with dangerous sequelae—both infectious risks and vascular injury or thromboses. Some of the most common features are described in Table 11-3.
Table 11-3 Common RBC Hemolytic Disorders by Predominant Morphology*
| Spherocytes |
| Bizarre Poikilocytes |
| Elliptocytes |
| Spiculated or Crenated RBCs |
| Prominent Basophilic Stippling |
| Irreversibly Sickled Cells |
| Intraerythrocytic Parasites |
| Target Cells |
| Nonspecific or Normal Morphology |
Hgb, hemoglobin; HMP, hexose-monophosphate shunt; RBC, red blood cell.
* Nonhemolytic disorders of similar morphology are enclosed in parentheses for reference.
† Usually associated with a positive Coombs test.
‡ Disease sometimes associated with this morphology.
Modified from Nathan DG, Oski FA, editors: Hematology of infancy and childhood, ed 2, Philadelphia, 1981, WB Saunders.
Hereditary spherocytosis (HS) is the most common cause of genetically determined hemolytic anemia in the white population. HS is transmitted frequently as an autosomal dominant trait, and it is named for the peculiar appearance of the RBCs on the peripheral blood smear (Fig. 11-13). The RBC membrane defect, which most commonly results from inherent cytoskeletal membrane instability due to spectrin deficiency, leads to loss of membrane. Membrane repair occurs, which decreases the normal RBC surface-to-volume ratio; this causes the normal biconcave disk to assume a more geometrically efficient spherical shape (Fig. 11-14). The new morphology creates a less pliable cell. The inability of this new cell to deform during transit through the splenic microcirculation leads to RBC destruction (Fig. 11-15). Osmotic fragility testing, used commonly in patients with suspected hereditary spherocytosis, is an excellent confirmatory test but is not pathognomonic for the diagnosis.
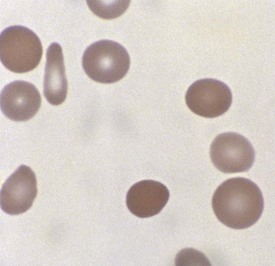
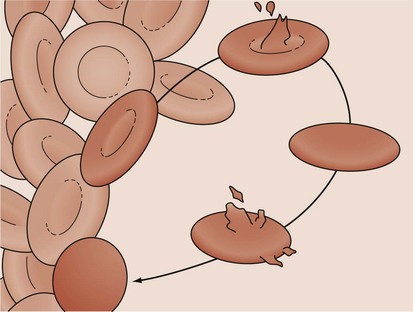
Figure 11-14 A developing spherocyte resulting from the process of repeated membrane fragmentation, loss, and repair.
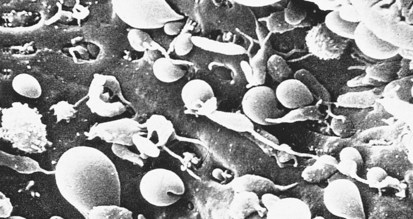
Hereditary elliptocytosis (HE) is another membrane defect morphologically distinct from hereditary spherocytosis (Fig. 11-16). However, its inheritance and pathophysiology of RBC destruction are similar to that in spherocytosis. In most instances, HE is a mild, well-compensated hemolytic anemia that is clinically insignificant unless splenic hypertrophy develops from another disease process. The elliptocyte form bears only a superficial similarity to that of the ovalocyte of iron deficiency anemia. Unlike those cells, the elliptocytes of HE have normal size, have a normal MCV, and are true elliptocytes. Elliptocytes may also be found in the peripheral blood smear of thalassemia or in megaloblastic anemia—although these findings do not suggest mixed pathologies.
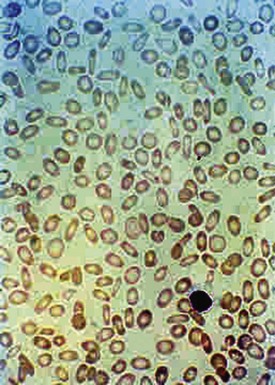
A number of hemolytic anemias are characterized by spiculated RBCs referred to as acanthocytes or echinocytes (Fig. 11-17). Abetalipoproteinemia generally presents as a neurologic disorder with progressive ataxia, retinitis pigmentosa, fat malabsorption, and the absence of chylomicrons and very low–density and low-density lipoproteins. Acanthocytes develop as a direct result of the alterations of the serum lipids. Altered membrane lipid composition changes the fluidity of the RBC, leading to the abnormal form. Malabsorption of fat-soluble vitamins in abetalipoproteinemia results in vitamin E deficiency, leaving the RBCs subject to oxidative injury. However, despite altered membrane fluidity and vitamin E deficiency, hemolysis in abetalipoproteinemia is mild.
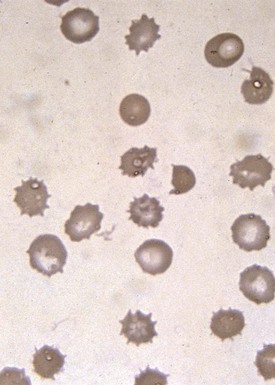
Target cells draw their name from their characteristic appearance on the peripheral blood smear. They are often seen as a secondary response to a process that increases the RBC membrane or decreases the RBC content, leading to an increase in the surface-to-volume ratio of the RBCs (Fig. 11-18, A). In a dried smear, the excess surface accumulates and bulges outward in the area that is normally the RBC’s central pallor, producing the characteristic target cell morphology. Liver diseases of any type, particularly obstructive hepatopathy, are well-known causes of target cell formation. Splenectomy decreases reticuloendothelial remodeling of reticulocytes, removes lipid-loaded RBC membranes, and leads to targeted RBCs. Mechanisms previously discussed that decrease RBC intracellular content, such as hypochromic, microcytic anemia, also induce target formation. In addition, target cells (or more accurately, pseudotarget cells) occur with various hemoglobinopathies. This happens more often in conditions associated with Hgb C, but also with Hgb S, D, and E (Figs. 11-19 and 11-20), because of aggregation of hemoglobin in the central region of the RBC (Fig. 11-18, B).
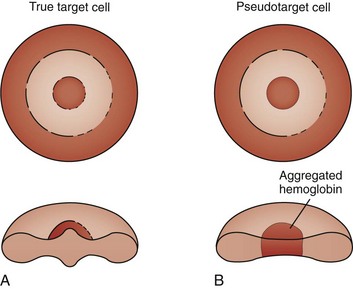
Figure 11-18 A, Schematic of the morphology of a target cell. B, Schematic of the morphology of a pseudotarget cell.
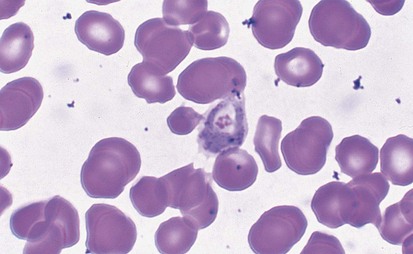
Malaria is the most frequent cause of hemolysis on a worldwide scale. The patient who contracts the disease after being fed on by the tropical Anopheles mosquito is parasitized within the RBCs with organisms at the merozoite stage (Fig. 11-21). The parasitization causes a clinical picture of intermittent fever, chills, and jaundice and may lead to encephalopathy, massive hemolysis with hemoglobinuria (blackwater fever), and death. The cause of the hemolysis has been attributed to multiple mechanisms including altered RBC osmotic fragility, membrane loss of negative surface charge, direct injury by the parasite, autoimmunity, splenic pitting, and hypersplenism.
Abnormalities of RBC enzymes may also lead to hemolysis. Although almost any RBC enzymes involved in RBC glycolysis or free radical detoxification via the pentose shunt may be responsible for hemolysis, glucose-6-phosphate dehydrogenase (G6PD) deficiency is by far the most frequent. A second enzyme, pyruvate kinase (PK), when deficient, also leads to a hemolytic state. The blood smears of patients with RBC enzyme deficiency–induced hemolysis are often normal, although occasionally with G6PD deficiency a suspicious morphology may be present (Fig. 11-22). More than 370 variants of G6PD have been identified, for which enzyme activity may be normal, elevated, or severely deficient. G6PD deficiency is transmitted by a sex-linked recessive mode of inheritance. Heterozygous females may rarely be affected because of inactivation of the X chromosome according to the Lyon hypothesis or because they are doubly heterozygous. Clinical syndromes may vary as well, with the degree of hemolysis inversely paralleling the level of G6PD activity.
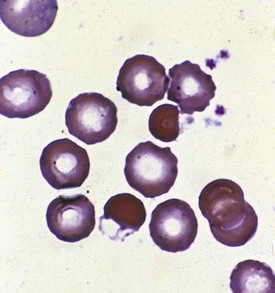
In all instances the hemolysis is due to the intracellular generation of free radicals and peroxides, which fail to be detoxified by the patient with G6PD deficiency. Chronic hemolysis is extremely mild and subclinical in the common types. Triggered hemolysis associated with G6PD deficiency (Mediterranean type) is severe and abrupt and parallels inversely the severe enzyme deficiency. In contrast, triggered hemolysis with G6PD deficiency (A type) may be severe but self-limited when the enzyme deficiency is mild. In addition, some agents that trigger hemolysis with G6PD deficiency (Mediterranean type) may be tolerated by patients with G6PD deficiency (A type). A list of some drugs associated with clinically significant hemolysis in G6PD deficiency is provided (Table 11-4). Research has deemphasized the role of certain drug triggers. In many instances the infection being treated with certain drugs, rather than the drugs themselves, is responsible for the hemolysis.
Table 11-4 Drugs Associated with Clinically Significant Hemolysis in Glucose-6-phosphate Dehydrogenase Deficiency
| Antimalarials |
| Antipyretics and Analgesics |
| Sulfa Drugs |
| Miscellaneous |
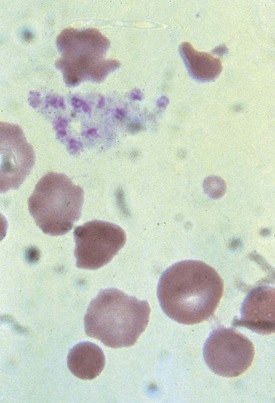
Direct and indirect Coombs tests, which evaluate the patient’s blood for the presence of anti-RBC antibody and complement on RBCs or in the serum, respectively, identify immune-mediated hemolytic anemia. Antibody- and complement-mediated RBC destruction produces spherocytes on the peripheral blood smear (Fig. 11-23). Coombs-positive hemolytic anemia in the newborn most often represents an isoimmune hemolytic anemia. This is caused by a maternal antibody that has crossed the placenta into the neonate, hemolyzing the newborn RBCs. Maternal antibodies form to fetal RBC antigens when fetal blood gains entry into the maternal circulation via a break in placental integrity. Maternal antibodies then cross the placenta and cause fetal RBC hemolysis. In the majority of cases of isoimmune hemolytic anemia, the maternal antibody is directed at ABO or Rh RBC antigens. When Rh antigen is the antibody target, the blood smear does not show spherocytes.
Hemoglobinopathies
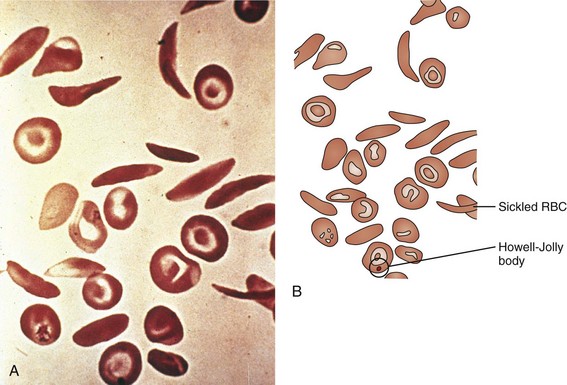
Of the hemoglobinopathies that have altered hemoglobin solubility, none is as well known or as ubiquitous as sickle cell disease. Substitution of valine for glutamic acid at position 6 in the β chain of the hemoglobin molecule leads to the cross-linking of one β chain to a second hemoglobin molecule’s β chain when the hemoglobin is in its deoxygenated state. This cross-linkage tips the solubility balance, leading to sickling of the RBC (see Fig. 11-19). Sickle cell disease is by no means exclusive to patients of African origin, with Mediterranean and Middle Eastern peoples also being affected. Sickle cell disease is an autosomal recessive disorder with the heterozygote having a significant but less than 50% proportion of hemoglobin of the sickle cell type. Heterozygous carriers of the sickle cell gene, although suffering a number of difficulties (e.g., poor urine-concentrating ability, occasional episodes of renal papillary necrosis with hematuria), have normal life expectancies and are virtually free of any significant consequences of their heterozygous state.
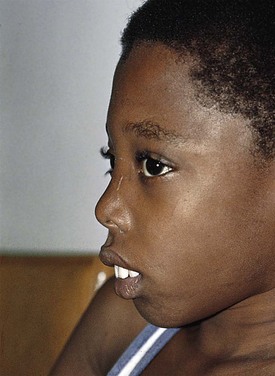
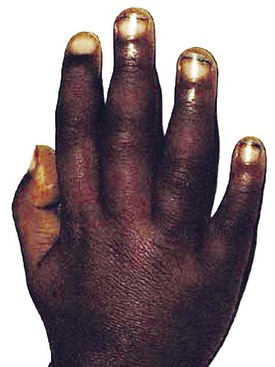
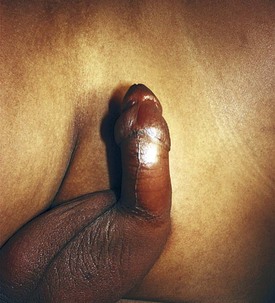
The clinical signs and symptoms of sickle cell anemia are due to decreased survival and altered rheology of the sickled RBC. As with any chronic hemolytic state, marrow cavity enlargement occurs, leading to maxillary hyperplasia and the so-called sickle cell facies (Fig. 11-24). Sickle cells have altered pliability and lose the ability to deform in the microcirculation (see Fig. 11-15). This leads to tissue infarction and subsequent pain. These vaso-occlusive crises, when occurring in certain locations, give rise to clinical manifestations such as dactylitis (Fig. 11-25), priapism (Fig. 11-26), splenic sequestration, and skin ulceration. The vaso-occlusive phenomenon may also lead to life-threatening complications of sickle cell disease: overwhelming infection with encapsulated organisms (most often Pneumococcus), acute chest syndrome, and stroke. The increased infectious risk in the sickle cell patient has multiple mechanisms, but by far the most important is splenic dysfunction related to congested blood flow and then, ultimately, splenic infarction.
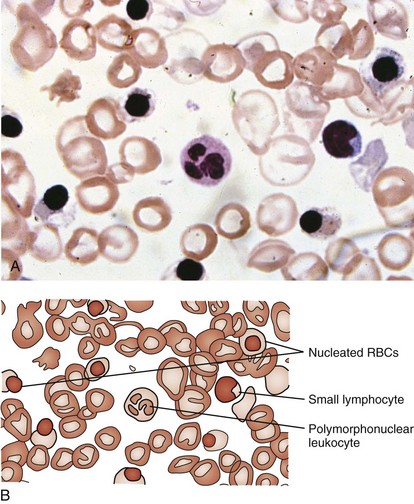
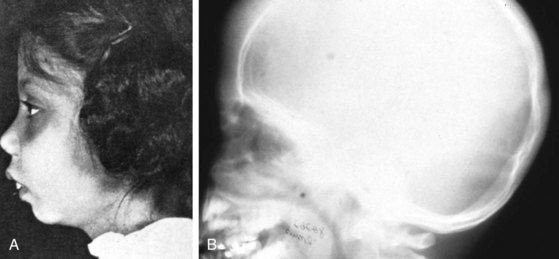
β-Thalassemia major (Cooley anemia) causes hypochromic, microcytic anemia that results from ineffective erythropoiesis caused by an imbalance between α- and β-hemoglobin chain synthesis. The peripheral blood smear shows hypochromia; microcytosis; target cells; basophilic stippling; and, often, a large number of nucleated RBCs (Fig. 11-27). β-Thalassemia major is associated with increased marrow activity, which is ineffectively attempting to correct the degree of anemia. The increased marrow activity expands the marrow cavity, producing a characteristic bony hyperplasia evidenced by physical and radiographic findings (Fig. 11-28). Untreated patients with thalassemia major have chronic and severe anemia, marked hepatosplenomegaly, scleral icterus, and listlessness and may have high-output cardiac failure secondary to severe anemia. In addition, malocclusion may occur because of molar hypertrophy. This picture is not usually confused with thalassemia trait, iron deficiency, or lead poisoning.
Although less common than sickle cell disease, anemia associated with hemoglobin C is not rare in the African-American population. As in sickle cell disease, Hgb C disease occurs because of one amino acid change. The change, again like Hgb S, is at the sixth position of the β chain but the replacement is a lysine rather than a valine. In its homozygous form, Hgb C disease is a mild disorder characterized by hemolytic anemia and splenomegaly. The tendency of Hgb C to aggregate into precipitates is responsible for the characteristic target (actually pseudotarget; see Fig. 11-18, B) morphology of the Hgb C homozygous and Hgb C trait (heterozygous) cells on the peripheral blood smear. Vaso-occlusive phenomena are not associated with this disease, although target cells are formed on the dried peripheral blood smear. However, Hgb C, when paired in a double heterozygous state with Hgb S, results in Hgb SC disease (see Fig. 11-20) and is associated with vaso-occlusive phenomena, although less severe than Hgb SS.
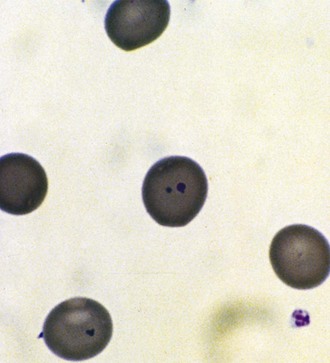
Unstable hemoglobin variants, unlike most hemolytic hemoglobinopathies, rarely have a characteristic morphology, although basophilic stippling and Heinz bodies may be noted on special staining of the peripheral blood (Fig. 11-29). The Heinz bodies represent hemoglobin aggregates that have precipitated intracellularly. RBCs in Heinz body anemia usually are normocytic but may be hypochromic as a result of RBC splenic “pitting” of precipitated hemoglobin. Because the precipitated hemoglobin may be mistaken for reticulum, reticulocyte counts may be spuriously high. Methylene blue staining of RBCs after they have incubated for a few hours can demonstrate the Heinz bodies. Unstable hemoglobinopathies have an autosomal dominant pattern of inheritance, and affected individuals are heterozygotes. A homozygous state would, in most cases, be incompatible with life. Congenital Heinz body hemolytic anemia is an important cause of congenital hemolytic anemia. This condition, although frequently a persistent process in the older infant, has been observed to resolve.
The Coagulation System
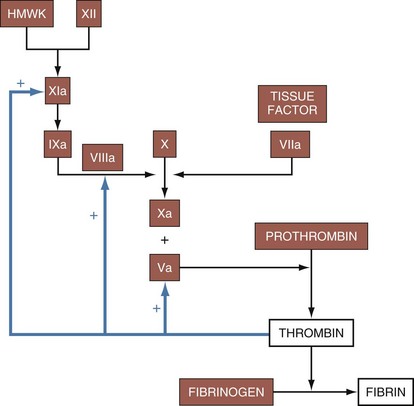
Platelets mature from megakaryocytes and have a life span of approximately 7 to 10 days. Platelets adhere to the site of injury by connecting to exposed von Willebrand factor on the vessel basement membrane. Subsequently, fibrinogen binding acts as a stimulus for the aggregation of other platelets. Once platelets have gathered at the site of injury, phospholipids on their surface interact with calcium to enable the hemostatic activity of the coagulation cascade. This process is a complex, interconnected series of reactions between enzymes and cofactors that leads to the deposition of fibrin (Fig. 11-30). The extrinsic pathway is activated when exposed tissue factor is contacted by circulating factor VIIa, causing the initial burst of thrombin production. The intrinsic pathway, through feedback mechanisms triggered by thrombin generation, plays an important role in clot propagation and continued fibrin production. Disorders of platelet plug formation commonly present with mucocutaneous oozing or bleeding, whereas disorders of coagulation factors often present with delayed and deeper tissue bleeding due to the ineffective fibrin deposition.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


