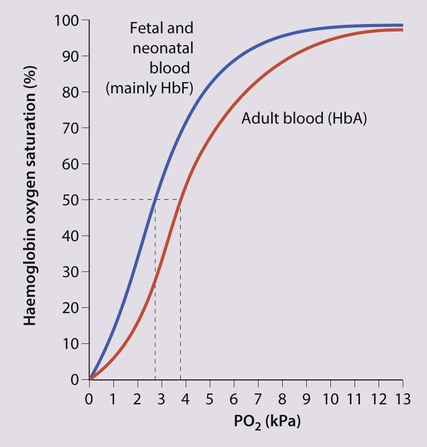
The most important difference between haemopoiesis in the fetus compared with postnatal life is the changing pattern of haemoglobin (Hb) production at each stage of development. The composition and names of these haemoglobins are shown in Table 22.1. Understanding the developmental changes in haemoglobin helps to explain the patterns of abnormal haemoglobin production in some inherited childhood anaemias. Embryonic haemoglobins (Hb Gower 1, Hb Gower 2 and Hb Portland) are produced between 4 and 8 weeks’ gestation, after which haemoglobin production switches to fetal haemoglobin (HbF). HbF is made up of 2 α chains and 2 γ chains (α2γ2) and is the main Hb during fetal life. HbF has a higher affinity for oxygen than adult Hb (HbA), and is therefore better able to hold on to oxygen, an advantage in the relatively hypoxic environment of the fetus (Fig. 22.1). At birth, the types of Hb are: HbF, HbA and HbA2. HbF is gradually replaced by HbA and HbA2 during the first year of life. By 1 year of age, the percentage of HbF is very low in healthy children and increased proportions of HbF are a sensitive indicator of some inherited disorders of haemoglobin production (haemoglobinopathies) . Table 22.1 Embryonic, fetal and adult haemoglobins • At birth, the Hb in term infants is high, 14–21.5 g/dl, to compensate for the low oxygen concentration in the fetus. The Hb falls over the first few weeks, mainly due to reduced red cell production, reaching a nadir of around 10 g/dl at 2 months of age (Fig. 22.2). Normal haematological values at birth and during childhood are shown in the Appendix. • Preterm babies have a steeper fall in Hb to a mean of 6.5–9 g/dl at 4–8 weeks chronological age. • Normal blood volume at birth varies with gestational age. In healthy term infants the average blood volume is 80 ml/kg; in preterm infants the average blood volume is 100 ml/kg. • Stores of iron, folic acid and vitamin B12 in term and preterm babies are adequate at birth. However, in preterm infants, stores of iron and folic acid are lower and are depleted more quickly, leading to deficiency after 2–4 months if the recommended daily intakes are not maintained by supplements. • White blood cell counts in neonates are higher than in older children (10–25 × 109/L). • Platelet counts at birth are within the normal adult range (150–400 × 109/L). Anaemia results from one or more of the following mechanisms: • Reduced red cell production – either due to ineffective erythropoiesis (e.g. iron deficiency, the commonest cause of anaemia) or due to red cell aplasia • Increased red cell destruction (haemolysis) There may be a combination of these three mechanisms, e.g. anaemia of prematurity. Using this approach, the principal causes of anaemia are shown in Figure 22.3 and a diagnostic approach to identifying their causes is shown in Figure 22.4. Diagnostic clues to ineffective erythropoiesis are: • Abnormal mean cell volume (MCV) of the red cells: low in iron deficiency and raised in folic acid deficiency. The main causes of iron deficiency are: Inadequate intake of iron is common in infants because additional iron is required for the increase in blood volume accompanying growth and to build up the child’s iron stores (Fig. 22.5). A 1-year-old infant requires an intake of iron of about 8 mg/day, which is about the same as his father (9 mg/day) but only half that of his mother (15 mg/day). • Breast milk (low iron content but 50% of the iron is absorbed) • Infant formula (supplemented with adequate amounts of iron) • Cow’s milk (higher iron content than breast milk but only 10% is absorbed) • Solids introduced at weaning, e.g. cereals (cereals are supplemented with iron but only 1% is absorbed). Iron deficiency may develop because of a delay in the introduction of mixed feeding beyond 6 months of age or to a diet with insufficient iron-rich foods, especially if it contains a large amount of cow’s milk (Box 22.1). Iron absorption is markedly increased when eaten with food rich in vitamin C (fresh fruit and vegetables) and is inhibited by tannin in tea. Most infants and children are asymptomatic until the Hb drops below 6–7 g/dl. As the anaemia worsens, children tire easily and young infants feed more slowly than usual. The history should include asking about blood loss and symptoms or signs suggesting malabsorption. They may appear pale but pallor is an unreliable sign unless confirmed by pallor of the conjunctivae, tongue or palmar creases. Some children have ‘pica’, a term which describes the inappropriate eating of non-food materials such as soil, chalk, gravel or foam rubber (see Case History 22.1). There is evidence that iron deficiency anaemia may be detrimental to behaviour and intellectual function. There are three main causes of red cell aplasia in children: • Congenital red cell aplasia (‘Diamond–Blackfan anaemia’) • Transient erythroblastopenia of childhood • Parvovirus B19 infection (this infection only causes red cell aplasia in children with inherited haemolytic anaemias and not in healthy children). The diagnostic clues to red cell aplasia are: • Red cell membrane disorders (e.g. hereditary spherocytosis) • Red cell enzyme disorders (e.g. glucose-6-phosphate dehydrogenase deficiency) • Haemoglobinopathies (abnormal haemoglobins, e.g. β-thalassaemia major, sickle cell disease). Haemolysis from increased red cell breakdown leads to: The diagnostic clues to haemolysis are: • Raised reticulocyte count (on the blood film this is called ‘polychromasia’ as the reticulocytes have a characteristic lilac colour) • Unconjugated bilirubinaemia and increased urinary urobilinogen • Abnormal appearance of the red cells on a blood film (e.g. spherocytes, sickle shaped or very hypochromic) (Fig. 22.6) • Positive direct antiglobulin test (only if an immune cause, as this test identifies antibody-coated red blood cells) • Jaundice – usually develops during childhood but may be intermittent; may cause severe haemolytic jaundice in the first few days of life • Anaemia – presents in childhood with mild anaemia (haemoglobin 9–11 g/dl), but the haemoglobin level may transiently fall during infections • Mild to moderate splenomegaly – depends on the rate of haemolysis • Aplastic crisis – uncommon, transient (2–4 weeks), caused by parvovirus B19 infection These are red blood cell disorders which cause haemolytic anaemia because of reduced or absent production of HbA (α- and β-thalassaemias) or because of the production of an abnormal Hb (e.g. sickle cell disease). α-Thalassaemias are caused by deletions (occasionally mutations) in the α-globin gene. β-Thalassaemia and sickle cell disease are caused by mutations in the β-globin gene. Clinical manifestations of the haemoglobinopathies affecting the β-chain are delayed until after 6 months of age when most of the HbF present at birth has been replaced by adult HbA (Fig. 22.7, Table 22.2). Table 22.2 Haemoglobins in haemoglobinopathies
Haematological disorders
Haemoglobin production in the fetus and newborn
Haemoglobin type
Globin chains
α-gene cluster
β-gene cluster
Embryonic
Hb Gower 1
ξ2
ε2
Hb Gower 2
α2
ε2
Hb Portland
ξ2
γ2
Fetal
HbF
α2
γ2
Adult
HbA
α2
β2
HbA2
α2
δ2
Haemoglobin types in newborns and adults
Newborn
HbF 74%, HbA 25%, HbA2 1%
Children >1 year old and adults
HbA 97%, HbA2 2%
Haematological values at birth and the first few weeks of life
Anaemia
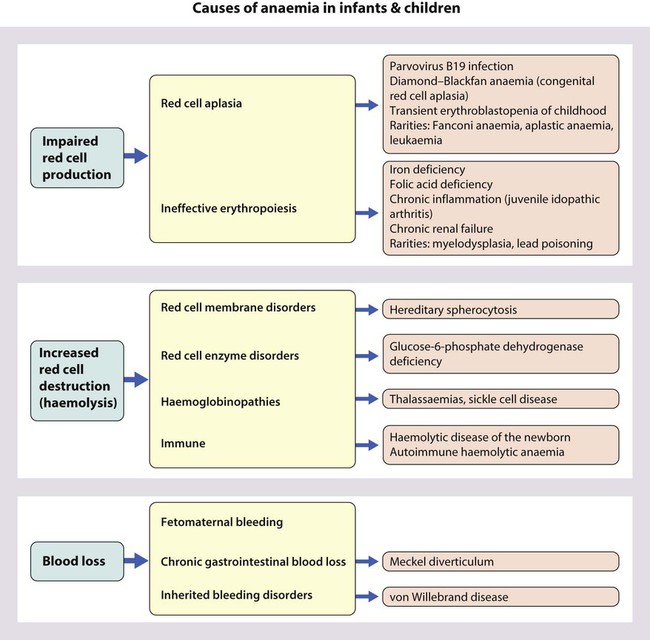
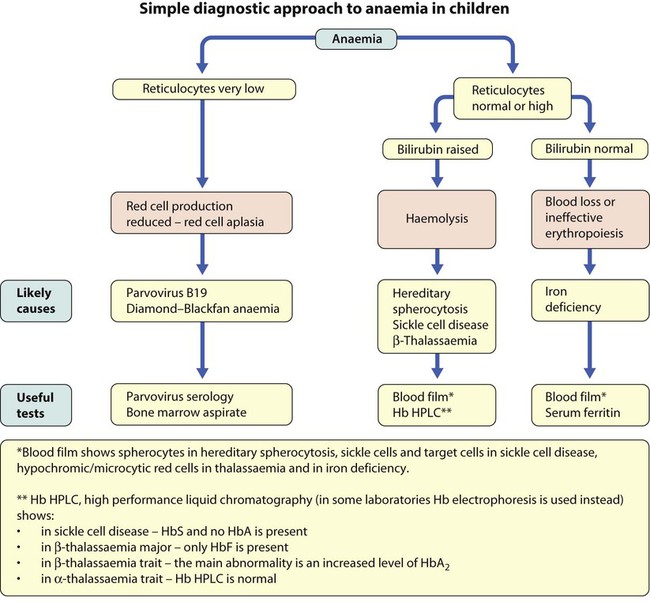
Causes of anaemia in infants and children
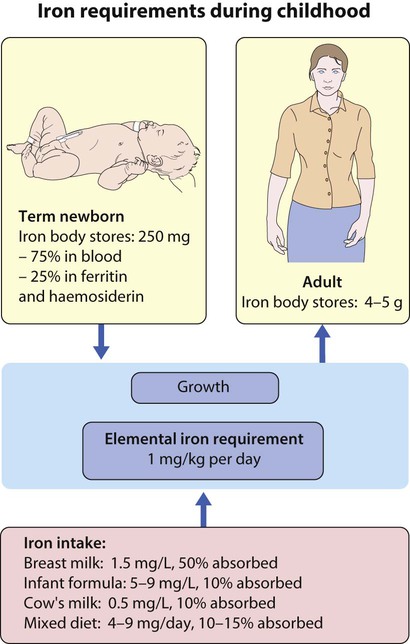
Iron deficiency
Clinical features
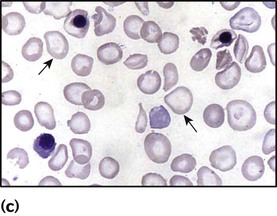
Red cell aplasia
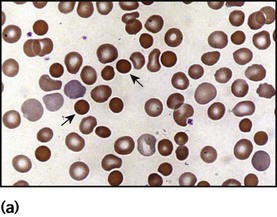
Increased red cell destruction (haemolytic anaemia)
Hereditary spherocytosis
Clinical features
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
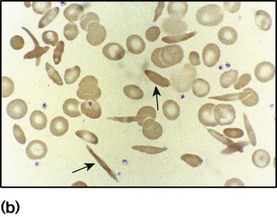
Haemoglobinopathies
HbA
HbA2
HbF
HbS
Newborn
25%
1%
74%
−
Adult
97%
2%
−
−
β-Thalassaemia trait
>90%
↑
+ ↑
−
β-Thalassaemia major
−
↑
↑
−
Sickle cell trait
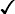
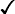
+ ↑
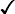
Sickle cell disease
−
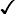
+ ↑
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


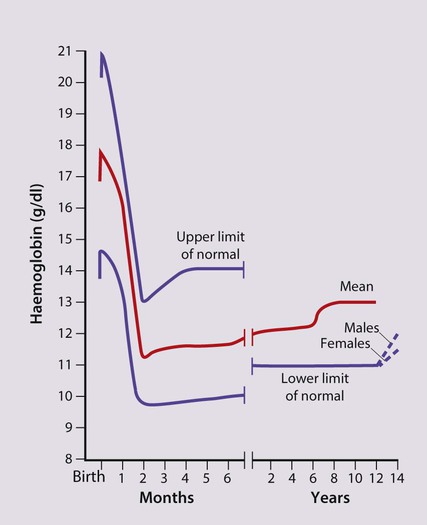
 The definition of anaemia varies with age: Hb <10 g/dl in infants (post-neonatal), Hb <11 g/dl from 1 to 12 years old.
The definition of anaemia varies with age: Hb <10 g/dl in infants (post-neonatal), Hb <11 g/dl from 1 to 12 years old. Infants should not be fed unmodified cow’s milk as its iron content is low and poorly absorbed.
Infants should not be fed unmodified cow’s milk as its iron content is low and poorly absorbed. Treatment of iron deficiency anaemia is with dietary advice and oral iron therapy for several months.
Treatment of iron deficiency anaemia is with dietary advice and oral iron therapy for several months.