Fig. 12.1
The GH/IGF-1 axis and its role in linear growth. The hypothalamic release of growth hormone releasing hormone (GHRH) stimulates the pulsatile release of growth hormone (GH) from the pituitary. The GH cell surface receptor (GHR) is widely expressed throughout the body. GH binds to the extracellular domain of GHR, inducing the upregulation of various anabolic target genes including insulin-like growth factor-1 (IGF-1). The majority of circulating IGF-1 forms a ternary complex with acid-labile subunit (ALS) and insulin-like growth factor binding protein-3 (IGFBP-3). IGF-1 acts in both an “endocrine fashion” (process A) and “autocrine/paracrine” fashion (process B). In addition to upregulating IGF-1 production, GH contributes directly to linear growth by inducing differentiation of the precursor cells within the growth plate towards chondrocytes (C). IGF-1 stimulates mitosis of epiphyseal chondrocytes (D) and also mediates the negative feedback of GH (E)
Growth Hormone and IGF-1
The precise mechanism by which GH is released and subsequently stimulates the release of IGF-1 is now well established [11–15] (Fig. 12.2). In humans, the majority of circulating IGF-1 is synthesized in the liver, although a low level of GH-dependent and GH-independent IGF-1 expression does occur in extra-hepatic tissues.
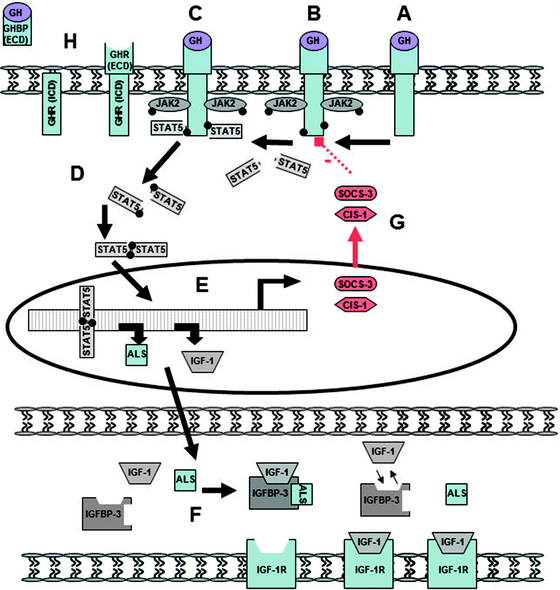
Fig. 12.2
The growth hormone receptor and JAK2/STAT5 signaling pathway. (A) Within its various target tissues, GH binds to the extracellular domain of the Growth Hormone Receptor (GHR), (B) inducing the intracellular auto-phosphorylation of Janus kinase 2 (JAK2). (C) In turn, phosphorylated JAK2, in association with activated GHR, leads to the phosphorylation of signal transducer and activator of transcription protein 5 (STAT5). (D) Activated STAT5 dimerizes and then (E) translocates to the nucleus, resulting in the upregulation of various anabolic target genes including IGF-1 and acid labile subunit (ALS) [12–14]. (F) IGF-1 and ALS pass to the circulation and form ternary complexes with insulin-like growth factor binding protein (IGFBP), about 75% as a 150-kDa complex with IGFBP-3. (G) Suppressors of cytokine signaling (SOCS) proteins are postreceptor inhibitors of cell signaling that mediate their effect via the JAK/STAT pathway [15]. GH rapidly and prominently induces expression of SOCS-3 and cytokine-inducible SH2-containing protein-1 (CIS-1) within the liver as part of a negative feedback loop that functions by blocking the phosphorylation of STAT5. SOCS-3 inhibits JAK2 by a mechanism requiring GHR. (H) The GHR has both an intra- and extracellular domain (ICD and ECD). Growth hormone receptor binding protein (GHBP), present within the circulation, is produced by the inducible metalloproteolytic cleavage of the GHR’s extracellular domain. Serum concentrations of this protein are thought to reflect GHR density [170]
Gender Differences in the GH/IGF-1 Pathway
GH is released in a pulsatile pattern that is gender specific, with males experiencing higher peaks and deeper troughs compared to females [16]. Interestingly, STAT5 exists in two genetically distinct, although highly homologous, forms (STAT5A and STAT5B) [17] which are known to differ somewhat in their tissue distribution [18]. Of note, while STAT5A and STAT5B are both required for normal GH-dependent growth, STAT5B is responsive to pulsatile GH whereas STAT5A is not. Indeed, STAT5B deficient male mice have pronounced growth impairment and tend to grow at a rate similar to normal females. Thus, the complex regulation of sexually dimorphic growth appears to be mediated, at least in part, by STAT5B “interpreting” the differing GH pulsatile secretion patterns of males vs. females [17]. Given this, it seems plausible that any interference within the GH/STAT5B/IGF-1 pathway is likely to have a more pronounced effect on growth patterns in males than females.
Insulin-Like Growth Factor Binding Proteins
The bioavailability of IGF-1 depends on its unbound or “free” fraction. Six specific high-affinity IGF-1 binding proteins (IGFBP-1 to IGFBP-6) are present within the circulation and can each bind IGF-1 with an affinity at least equal to the binding of IGF-1 to the IGF receptor [19]. The insulin-like growth factor binding proteins (IGFBPs) are each regulated by specific proteases that dramatically reduce their IGF-1 binding affinity. The specific function and structure of the six IGFBPs differ significantly [20]. IGFBP-1, -2, -4, and -6 primarily inhibit IGF-1 by tightly binding to it and preventing it from binding to its receptor [19, 21, 22]. Conversely, IGFBP-3 potentiates the action of IGF-1 by “loosely” binding to it, thus prolonging the time it is available within the circulation to interact with its receptor. About 75% of IGF-1 circulates as a 150-kDa ternary complex composed of IGF-1, acid-labile subunit (ALS), and IGFBP-3 [19]. This large complex, which cannot cross the endothelial barrier [23] significantly increases the half-life of IGF-1 from less than 10 min to greater than 16 h. Caloric and protein restriction can cause a reduction in the levels of IGFBP-3 [24, 25].
Growth Plate Proliferation, Senescence, and Fusion
The normal age-dependent decline in growth rate is due primarily to a senescent decline in the rate of growth plate chondrocyte proliferation [26, 27] referred to as “growth plate senescence” [28–30]. The proliferative capacity of the “stem-like” cells within the resting zone of the growth plate is finite. Thus, “senescence” is not a function of time per se, but of proliferative cycle number. Given this, it becomes apparent that interventions that slow the proliferation rate of growth plate chondrocytes, such as glucocorticoid exposure, will also slow the rate of growth plate senescence [29, 31]. That is to say, following transient growth inhibition, growth plates are “less senescent,” retaining a greater proliferative capacity than expected for age. Thus, in the “post inhibitory period,” the growth plate will show a greater growth rate than expected for age, resulting in “catch-up growth,” the apparently “accelerated” linear growth that occurs after resolution of a growth-inhibiting condition [30, 32].
The pubertal growth spurt is primarily induced by estrogen, which acts to increase the activity of the GH/IGF-1 axis [33, 34]. In addition, the sex steroids, especially the androgens, appear to stimulate growth by a direct effect on growth plate chondrocytes [35–37]. Estrogen is also known to be the key hormone that promotes epiphyseal fusion [28].
Monitoring and Assessment of Growth
Standardized charts are available for graphically recording height, weight, and height velocity such that an individual child’s growth can be compared to normative values [38–40]. Wherever possible, reference data most appropriate to the child being monitored should be utilized. An individual child’s growth measurement can be represented as a percentile or as a standard deviation score (SDS), a quantitative expression of distance from the reference population mean (50th percentile) for the same age and gender [41]. Healthy children grow steadily along the same height percentile and hence maintain the same SDS for height from early childhood through until adulthood. Combined parental heights can be used to estimate a child’s potential height [41]. Some temporary deviation from the usual growth channel may occur if the pubertal growth spurt occurs particularly early (temporary increase in height velocity and height centiles) or late (temporary decrease in height velocity and height centiles).
Definitions of Impaired Growth
Within a large patient group, skewing of SDS for height below population reference values is evidence of disease-associated growth impairment. Mean height SDS of a population characterized by normal growth approximates zero. Growth disturbance in an individual child is indicated by an abnormal growth rate [41]. A definition in terms of static height measurement, although sometimes used, may be misleading, since it is so influenced by parental heights. An individual child may be normally short; conversely a previously tall child may not have increased his height in 2 years, but still be of average stature. A shift from higher to lower centiles on a growth chart of height attained more sensibly signifies growth faltering. Height velocity, expressed either as a centile or as a SDS for age and gender, is the most sensitive parameter by which to recognize impaired growth.
Growth in Pediatric IBD
Prevalence of Growth Impairment in IBD
Inflammatory disease occurring during early adolescence is likely to have a major impact on nutritional status and growth because of the very rapid accumulation of lean body mass that normally occurs at this time. Further, boys are more vulnerable to disturbances in growth than girls because their growth spurt comes later and is ultimately longer and greater [4, 42].
Crohn Disease
Several studies have characterized the growth of children with Crohn disease as treated in the 1980s and into the 1990s [1, 43–48]. These studies are important as a benchmark of outcomes with traditional therapy. It is to be hoped that the now better understanding of the pathogenesis of growth impairment, together with the greater efficacy of current therapeutic regimens in healing intestinal inflammation, may lead to enhanced growth of young patients diagnosed now.
As summarized in Table 12.1, the percentage of patients with Crohn disease, whose growth is affected, varies with the time of assessment, the definition of growth impairment, and with the nature of the population under study (tertiary referral center vs. population based) [1, 43–53]. It has nevertheless been consistently observed that impairment of linear growth is common prior to recognition of Crohn disease as well as during the subsequent years, and that height at maturity has often been compromised [1, 43–52]. It is also apparent that these problems are more frequent among males than females, independent of disease location or severity [1, 53–57]. The basis of this observed gender difference is yet to be elucidated. Interestingly, as the incidence of CD increases in geographic regions where it was previously rare, reports demonstrate that similar patterns of impaired growth are being observed [58, 59].
Table 12.1
Prevalence of linear growth impairment in pediatric Crohn disease. Varying definitions and times of assessment (at the time of diagnosis and during follow-up) are applied
Study details (ref) | Time of assessment | Patients studied | n | Definition of linear growth impairment | Percentage with growth impairment |
|---|---|---|---|---|---|
Baltimore, USA 1961–1985 [44] | At diagnosis | Prepubertal (Tanner I or II) | 50 | Decrease in height velocity prior to diagnosis | 88% |
Toronto, Canada 1980–1988 [1] | During follow-up | Prepubertal (Tanner I or II) | 100 | Height velocity ≤2 SD for age for ≥2 years | 49% |
Sweden 1983–1987 [45] | During follow-up | Population-based cohort <16 years at Dx | 46 | Height velocity ≤2 SD for age for 1 year | 65% |
New York, USA 1979–1989 [46] | At maturity | Children in tertiary care | 38 | Failure to reach predicted adult height | 37% |
Toronto, Canada 1990 to 1999 [48] | During follow-up | Prepubertal (Tanner I or II) | 161 | Height velocity ≤2 SD for age for ≥2 years | 54% |
United Kingdom 1998–1999 [49] | At diagnosis | Population-based cohort <16 years at Dx | 338 | Height SDS ≤1.96 | 13% |
Israel 1991–2003 [50] | At diagnosis | Children in tertiary care | 93 | Height SDS ≤2.0 | 20% |
France 1988–2004 [53] | At diagnosis | Population-based cohort <17 years at Dx | 261 | Height SDS ≤2.0 | 9.5% |
At the time of diagnosis mean SDS for height is reduced among children with Crohn disease as a group compared to reference populations (Table 12.2), an indication of the growth retardation occurring prior to recognition and treatment of intestinal inflammation [1, 44, 45, 48–50]. During the decade 1990–1999 in Toronto, mean SDS for height at time of diagnosis among 161 Tanner stage 1 or 2 children was −0.74 ± 1.2 [48], indicating overall lesser growth delay in comparison to the earlier decade [1]. Nevertheless the percentage of children with height less than the 5th centile (SDS score ≤1.8), based on Center for Disease Control 2000 data, was still 22% [48]. Mean SDS for height among 333 patients aged less than 16 years was −0.54 (95% CI −0.67 to −0.41) in a 1998–1999 population-based surveillance study of incident IBD in the United Kingdom [49]. Thirteen percent were below the third centile (SDS ≤1.96) for height based on data from Child Growth Foundation, London [49]. In Israel SDS for height at diagnosis among a cohort of 93 patients aged less than 18 years was −0.56 ± 1.16, but 20% had SDS ≤2.0 [50]. Taken together these data confirm that growth delay prior to diagnosis remains a challenge [48–50].
Table 12.2
Mean height standard deviation scores for height in children diagnosed with Crohn disease prior to or in early puberty (Tanner stage I or II)
Study (ref) | Patients studied | n | Mean height SDS (SD) | |
|---|---|---|---|---|
At diagnosis | At maturity | |||
Baltimore, USA 1961–1985 [44] | Prepubertal (Tanner I or II) | 50 | −0.48 | Not assessed |
Toronto, Canada 1980–1988 [1] | Prepubertal (Tanner I or II) | 100 | −1.1 (1.3) | −0.82 (1.1) |
Sweden 1983–1987 [45] | Population-based cohort <16 years at Dx | 46 | −0.5 (1.4) | −0.4 (1.1) |
Toronto, Canada 1990–1999 [48] | Prepubertal (Tanner I or II) | 161 | −0.74 (1.2) | −0.70 (1.2) |
United Kingdom 1998–1999 [49] | Population-based cohort <16 years at Dx | 338 | −0.54 | Not assessed |
Israel 1991–2003 [50] | Children in tertiary care | 93 | −0.56 (1.16) | Not assessed |
Leiden, Netherlands Reported in 2002 [52] | Children in tertiary care | 64 | Not reported | −0.9 (1.2) |
London, UK 1996–2002 [51] | Prepubertal children in tertiary care | 20 | Not reported | −0.57 (0.3) |
Delay in epiphyseal closure allows growth to continue longer than normal. Hence mean SDS for height may improve over the course of treatment, when the chronic inflammation can be controlled [1, 45, 48]. No population-based cohort studies have compared preillness height centiles with final adult stature in order to determine how often catch-up growth is complete. In spite of gains, mean adult height of patients with prepubertal onset of disease remains reduced compared to population reference data [1, 45, 48, 51, 52]. Studies suggesting otherwise have included patients with postpubertal onset of disease, and therefore not at risk for growth impairment [60].
Ulcerative Colitis
Cohort data are sparse in comparison to Crohn disease, but in general at diagnosis no significant reduction is observed in height-for-age SDS among young patients with ulcerative colitis compared to the reference population [45, 47, 49]. As an example, SDS for height was not reduced (mean −0.12, 95%CI −0.30 to 0.05) in 143 children and adolescents with incident UC in the British pediatric surveillance study [49].
In follow-up growth impairment remains a less frequent complication, although relatively few studies have carefully described linear growth in ulcerative colitis as compared to the abundance of studies in Crohn disease. Hildebrand et al. observed that 11 (24%) of 45 children had a height velocity ≤2.0 SD during at least 1 year [45]. Final attained mean height was comparable to reference population data in this study [45].
Why linear growth impairment is less common in ulcerative colitis than in Crohn disease is not entirely clear. The usual colitic symptom of bloody diarrhea is more promptly investigated than the often subtle presenting symptoms of Crohn disease, accounting at least in part for the lesser effect on growth prior to diagnosis. Disease-related differences in cytokine production are likely also important.
Pathophysiology of Growth Impairment in IBD
As summarized in Table 12.3, several interrelated factors contribute to linear growth impairment in children with IBD. The fundamental mechanisms have recently been comprehensively reviewed [61].
Table 12.3
Factors contributing to growth impairment in children with Crohn disease
Factor | Explanation |
|---|---|
Pro-inflammatory cytokines | Direct interference with IGF-1 mediation of linear growth |
Decreased food intake | Cytokine-mediated anorexia, fear of worsening gastrointestinal symptoms |
Stool losses | Mucosal damage leading to protein-losing enteropathy; diffuse small intestinal disease or resection leading to steatorrhea |
Increased nutritional needs | Fever; required catch-up growth |
Corticosteroid treatment | Interference with growth hormone and insulin-like growth factor-1 |
Chronic Caloric Insufficiency
Growth requires energy. Chronic undernutrition has long been implicated and remains an important and remediable cause of growth retardation [62]. Multiple factors contribute to malnutrition. However, reduced intake, rather than excessive loss or increased need, is generally the major cause of the caloric insufficiency. Kirschner et al. reported caloric intakes of growth-impaired patients to average 54% of that recommended for children of similar height age [63]. Food restriction may be deliberate to avoid symptoms. More importantly, cytokine-mediated disease-related anorexia may be profound. Work in a rat model of colitis suggests that tumor necrosis factor-alpha (TNF-α) interacts with hypothalamic appetite pathways [64]. While clinical studies have demonstrated that significant intestinal fat malabsorption is uncommon [65], leakage of protein is frequent [66]. In general, resting energy expenditure (REE) does not differ from normal in patients with inactive disease, but can exceed predicted rates in the presence of fever and sepsis [67]. Moreover, malnourished adolescents with CD fail to reduce their REE as efficiently as comparably malnourished patients with anorexia nervosa [67]. Reduction in REE is a normal biologic response to conserve energy. This relative failure of a compensatory mechanism has been attributed to the effects of pro-inflammatory cytokines.
Direct Cytokine Effects
A simple nutritional hypothesis, where adequate caloric delivery would remediate any growth impairment, fails to explain all the observations related to growth patterns among children with IBD. To date, a variety of cytokines have been implicated in the pathogenesis of IBD including TNF-alpha, interferon-gamma (IFN-gamma), and multiple interleukins (including IL-6, IL-12, IL-17, and IL-23). The direct growth-inhibiting effects of pro-inflammatory cytokines released from the inflamed intestine have been increasingly recognized [68–71].
Disruption of the GH/IGF-1 Axis
As described earlier, IGF-1, produced by the liver in response to GH stimulation, is the key mediator of GH effects at the growth plate of bones. An association between impaired growth in children with Crohn disease and low IGF-1 levels is well recognized [72]. However, GH production in this setting has been shown to be normal [73]. The molecular mechanisms by which cytokines induce this state of “GH resistance” have not yet been completely elucidated. Conceptually, they could involve downregulation of the GH receptor (GHR), upregulation of postreceptor inhibitory proteins, reduced protein synthesis, and/or increased protein degradation. Information from both animal models and/or human studies supports each of these potential mechanisms [14, 15, 68, 70, 74–86] (Fig. 12.3).
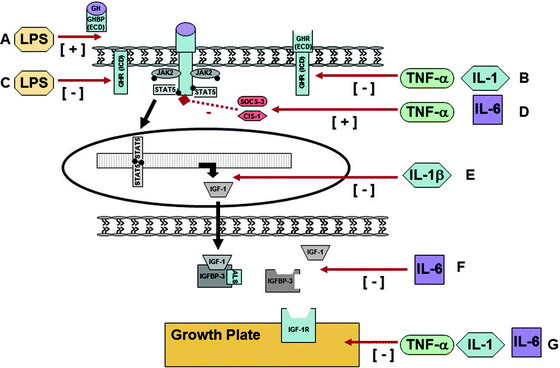
Fig. 12.3
Confirmed and potential molecular mechanisms that underpin the development of GH resistance in Crohn disease. At the growth hormone receptor: (A) Endotoxin exposure, specifically lipopolysaccharide (LPS), reduces GHR density by inducing GHR proteolysis and increasing the shedding of GHBP [74] (mechanism not yet ascertained). (B) TNF alpha has been demonstrated to downregulate GHR formation via inhibition of Sp1/Sp3’s ability to transactivate the GHR gene [75]. Il-1 suppresses GHR promoter activity [75]. (C) LPS can directly inhibit GHR gene expression via a cytokine-independent mechanism through the TLR-4/MD2 signaling pathway that results in a cytokine response, significant reduction in GHR promoter activity. Importantly, the addition of anti-TNF-alpha antibody failed to abrogate this effect [76]. Via postreceptor inhibitory proteins: (D) IL-6 and TNF-alpha can upregulate the expression of SOCS-3 and cytokine-inducible SH2-containing protein (CIS)1 [14, 77]. Both of these proteins have, in turn, been shown to inhibit GH signaling by blocking the phosphorylation of STAT5 [15, 78, 79]. Via reduced protein synthesis: (E) IL-1β has been shown to reduce IGF-1 mRNA levels. The mechanism is yet to be elucidated, but does not appear to be via upregulation of SOCS nor by impairment of JAK2/STAT5 signaling [80]. Via increased protein clearance: (F) IL-6 has been implicated in a reduction in IGFBP-3 levels due to either reduced production and/or increased proteolysis [81]. Previously, low levels of IGFBP-3 have been associated with accelerated clearance, and hence lower levels, of IGF-1 [81]. Via IGF-1 independent mechanisms: (G) Animal experiments have shown that TNF-α and interleukin-1 (IL-1) increase chondrocyte death and thus may have a deleterious effect on growth [70]. Cytokines appear to impair end-organ responsiveness to circulating testosterone [82]. IL-6 exposure promotes osteoclast maturation and activation, affects osteoblasts, is associated with osteoclast/osteoblast uncoupling, and results in thinning of the growth plate 68, [83–86]. Although the mechanism is yet to be determined, laboratory evidence suggests that it is independent of IGF-1 [83]
IGF-1 Independent Mechanisms
Inflammatory cytokines inhibit linear growth through pathways other than IGF-1 production. Animal experiments have shown that TNF-α and interleukin-1 (IL-1) increase chondrocyte death, and thus may have a deleterious effect on growth [70]. In an organ culture model of fetal rat parietal bone, marked impairment in osteoblast function and bone growth was observed with the addition of serum from children with CD, but not from children with ulcerative colitis, nor from healthy controls [71]. Finally, cytokines appear to impair end-organ responsiveness to circulating testosterone, thereby compounding the effects of under-nutrition in delaying progression through puberty [82].
The Role of IL-6 in Growth Impairment
As with a number of chronic inflammatory conditions, IL-6 is known to be elevated in the serum of pediatric patients with active CD and predictive of clinical relapse [87]. IL-6 activates STAT3 via the glycoprotein 130 signaling receptor (gp130); a process that is negatively regulated by SOCS-3 [88–90]. SOCS-3 is also a negative regulator of GH signaling. Very recently, it was confirmed that IL-6:STAT3 activation correlates with mucosal inflammation in active pediatric-onset CD [91, 92].
Transgenic mice with defective growth have been found to over-express interleukin-6 (IL-6). Antibody to IL-6 partially corrected the growth defect, whereas administration of IL-6 led to a decrease in IGF-1 before food intake was affected [68]. Similar to CD, children with juvenile idiopathic arthritis (JIA) also present with linear growth failure [93, 94]. Of note, IGF-1 levels are negatively correlated with IL-6 among this patient group [68]. The exact mechanism underpinning this observation, however, is not completely clear. While these, and other data [95], suggest an IL-6 mediated decrease in IGF-1 production [68]; work by DeBenedetti et al. suggests the primary mechanism is a reduction in IGFBP-3 levels due to reduced production and/or increased proteolysis of this binding protein [81]. Previously, low levels of IGFBP-3 have been associated with accelerated clearance, and hence low levels, of IGF-1 [81].
Recent studies in both of these pediatric patient groups have demonstrated a significant “uncoupling” of osteoblast and osteoclast activities [83, 96–98]. Concurrent mouse and human studies have shown that chronic IL-6 exposure promotes osteoclast maturation and activation, affects osteoblasts, is associated with osteoclast/osteoblast uncoupling, and results in thinning of the growth plate [68, 83–86]. Again, while the mechanism is yet to be determined, laboratory evidence suggests that it is independent of IGF-1 [83].
Taken together, these data suggest that increased IL-6 may represent a major generalized mechanism by which chronic inflammation affects the developing skeleton. This would imply that anti-IL6 therapeutic approaches, which have shown promising anti-inflammatory efficacy in CD, rheumatoid arthritis, and systemic JIA [99–102], may also specifically address the problem of growth impairment.
The Interplay Between Nutrition and Cytokines
The relative contributions of malnutrition and inflammation to linear growth delay were explored by Ballinger et al. using a rat model of TNBS colitis [69]. Two control groups were used: healthy controls with free access to food and a pair-fed group comprised of healthy animals with daily food intake restricted to match that of colitic rats [69]. In the colitic rats, IGF-1 levels were reduced to 35% of control values. Comparison with the healthy but undernourished pair-fed rats suggested that malnutrition accounted for 53% of the total depression of IGF-1 in colitic rats, with the remaining 47% attributable to inflammation [69].
Disruption of the GH/IGF-1 Axis by Cytokine-Independent Molecular Pathways
Impaired intestinal barrier function is a recognized feature in some patients with CD, and may predispose them to chronic, subclinical, endotoxin exposure, specifically lipopolysaccharide (LPS) [103]. Various groups are currently investigating whether LPS directly interferes with the GH/IGF-1 axis via cytokine-independent mechanisms. To date, in vivo data from a mouse model have demonstrated that LPS exposure reduces GHR density by inducing GHR proteolysis, probably via the metalloprotease cleavage site, resulting in the increased shedding of GHBP [74]. More recent in vitro data demonstrate that LPS can directly inhibit GHR promoter activity and subsequent expression through an effect on the TLR-4 signaling pathway [76]. Both mechanisms are seemingly independent of the inflammatory cytokine cascade and the addition of anti-TNF-alpha antibody failed to abrogate the effect [76]. Although intriguing, the clinical significance of these findings and their relative importance in the setting of growth impairment and CD are yet to be determined.
Corticosteroid Suppression of Linear Growth
The growth suppressive effects of glucocorticoids are multifactorial and can occur at virtually any point along the growth axis (Table 12.4) [104]. In general, exogenous corticosteroids are considered to create a state of functional GH deficiency [105]. Dose, preparation, and timing of glucocorticoids all influence the degree of growth suppression observed. It appears that concentrations of glucocorticoids required to exert direct suppression on the growth plate may be lower than those required to suppress GH secretion. Growth, particularly in prepubertal children, can be impaired by relatively modest daily doses of prednisone (3–5 mg/m2) [104]. This effect may be reduced, but is not necessarily eliminated, by alternate-day therapy. Selectively eliminating evening administration may avoid blunting of both nocturnal GH secretion and/or ACTH-induced adrenal androgen production [104]. Catch-up growth, following the cessation of glucocorticoid therapy, does not always fully compensate for growth deficits, particularly when treatment occurs during puberty. Although chronic daily dosing and frequent induction courses of steroids have been shown to lead to bone demineralization; at present there is not good evidence that short-term use of steroids for the induction of remission in CD is detrimental to long-term growth.
Table 12.4
The effects of exogenous glucocorticoid therapy related to linear growth (Adapted from Allen [104])
GH/IGF-1 axis |
Inhibit endogenous GH secretion |
Reduce pulsatile release of GH |
Increase somatostatin |
Interference with the GHR |
Reduce GHR expression |
Reduce GHR binding |
Uncouple GHR from signal transduction components |
Reduce IGF-1 activity levels |
Reduced activation of STAT5b |
Increased levels of IGFBP-3 |
Skeletal system |
Growth plate |
Inhibit chondrocyte mitosis |
Inhibit IGF-1 induced chondrocyte proliferation |
Inhibit epiphyseal maturation |
Skeletal matrix |
Diminish activity of enzymes required for posttranslational procollagen chain modification |
Inhibit collagen synthesis |
Increase collagen degradation |
Inhibit osteoblast function |
Peripheral tissues |
Calcium balance |
Decrease intestinal calcium absorption |
Increase urinary calcium excretion |
Body composition |
Increase protein catabolism |
Decrease lipid oxidation |
Inhibit secretion of adrenal sex steroids |
Reduce direct growth stimulatory effect of sex steroids |
Reduce usual augmentation of GH release |
The Pathogenesis of Pubertal Delay and its Influence on Growth Impairment
Puberty is frequently delayed in young patients with CD [105]. Similar to linear growth impairment, although undernutrition has been frequently considered the main reason for delayed puberty in children with CD, there is a group of patients with persistently active disease who do not enter puberty despite the provision of adequate energy [106]. Experimental colitis models demonstrate that inflammatory mediators potentiate the puberty-delaying effects of undernutrition [105] via alterations in gonadotropin releasing hormone (GnRH) secretion patterns, although which specific inflammatory cytokines impact on puberty is yet to be determined. However, both human and experimental data suggest that there is also an element of gonadotropin resistance in pubertal delay, and in vitro studies implicate TNF-alpha in the downregulation of androgen gene expression [107]. Although Cushing’s disease has been associated with pubertal delay [108], it is not known whether the doses of corticosteroid used in the management of CD are sufficient to delay either the onset or progression of puberty [105].
Influence of Genetic Factors
A number of genetic polymorphisms have been implicated in CD susceptibility and pathogenesis, the most prominent of which are within the NOD2 gene. While some investigators [109, 110] have suggested that CD-associated NOD2 polymorphisms may be determinants of growth impairment, neither analysis controlled for disease location. A subsequent careful analysis of growth prior to and following diagnosis found no such association [50]. Scottish pediatric data suggest an association between polymorphisms in the IBD5 susceptibility locus and low anthropometric centiles at diagnosis [111]. Similarly, data from Boston highlight a potential association with the CD susceptibility allele within ATG16L1 [112].
It is feasible that common genetic polymorphisms which alter cytokine expression may contribute to growth impairment but not influence overall susceptibility to CD. A recent study of Israeli patients suggests that relatively common variations in the promoter region for TNF-α may have an independent effect on linear growth outcomes [113]. Similarly, data from Sawczenko et al. demonstrate a potential causal relationship between variation in the promoter region for Il-6, subsequent IL-6 expression, and a differential in linear growth impairment during active inflammation [95]. Confirmation of these and similar findings is awaited, and may help better elucidate the complex molecular interactions pertinent to the pathophysiology of growth impairment.
Facilitation of Normal Growth in IBD
The Importance of Prompt Recognition of IBD
The clinical presentation of childhood Crohn disease may be subtle and varied. Impairment of linear growth and concomitant delay in sexual maturation may precede the development of intestinal symptoms and dominate the presentation. Prompt diagnosis is important in avoiding a long period of growth retardation. The greater the height deficit at diagnosis, the greater is the demand for catch-up growth.
The Importance of Monitoring Growth
In caring for children with IBD, it is important to obtain preillness and parental heights [114], so that the impact of the chronic intestinal inflammation can be fully appreciated. Following diagnosis and institution of treatment, regular measurement and charting of height, together with calculation of height velocity, are central to management. A properly calibrated wall-mounted stadiometer is required for accurate and reproducible serial measurements.
Part of the assessment of response to therapy in children with IBD is a regular analysis of whether rate of growth is normal for age and pubertal stage and whether catch-up growth to preillness centiles is being achieved. Height velocity should be appraised in the context of current pubertal stage, because of the variation in normal rates of growth before puberty, during puberty, and near the end of puberty. If growth and puberty appear either delayed or very advanced, radiologic determination of bone age can be used to indicate the remaining growth potential. Delayed radiological bone age suggests greater potential for catch-up growth than may be anticipated for the subject’s chronologic age. Conversely, in the subject with growth failure and a normal bone age, the potential window to achieve any growth catch-up may be very small.
One of the difficulties in evaluating growth in response to a therapy is the relatively long interval of time required for valid assessment. Published normal standards for height velocity throughout childhood are based on height increments during 12-month periods [115]. When growth velocity is calculated over short time periods, small errors in individual measurements are significantly magnified, and the normal seasonal variation in growth is overlooked. The consensus from pediatric endocrinologists is that height velocity should be calculated over intervals no shorter than 6 months [115]. On a research basis, efforts to reflect growth changes over intervals shorter than 6 months have focused on measuring changes in lower leg length by knemometry and on determination of circulating levels of markers of bone and collagen metabolism [115–117]. The clinical utility of routine serial assessment of the GH/IGF-1 axis is yet to be ascertained [118]. A valid indicator of contemporaneous linear growth would allow for a more timely change in therapy. A summary of techniques that should be employed to clinically assess and monitor linear growth through to adulthood, based on Heuschkel and colleagues management guideline, are presented in Table 12.5 [119].
Table 12.5
Techniques to assess and monitor linear growth in children with CD
Initial evaluation |
Accurate measurement of the patient’s height and weight by trained staff using reliable equipment |
Accurate pubertal assessment |
Accurate measurement of the biological parents’ heights and calculation of mid-parental height (MPH) |
Formula to estimate a subject’s potential adult height |
Male: MPH plus 6.5 cm; Female: MPH minus 6.5 cm
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|