Fig. 8.1
Stimulation and action of intrinsic GH. GH is secreted in the pituitary gland and stimulated by hypothalamic hormones, such as GHRH and ghrelin, and is suppressed by somatostatin. GH acts on many peripheral tissues and plays a role in linear growth, bone metabolism, adipose metabolism, protein metabolism, and saccarometabolism
GHRH selectively induces GH secretion from the pituitary gland through the GHRH receptor [3, 4]. Somatostatin suppresses GH pulse amplitude and frequency, and inhibits central GHRH release via direct synaptic connections with hypothalamic neurons, but does not affect GH biosynthesis [2]. Hypothalamic GHRH and somatostatin are secreted in independent waves and interact to generate pulsatile GH release together with additional GH secretagogues (GHS). Ghrelin binds to the GHS receptor to induce hypothalamic GHRH and pituitary GH [5, 6]. The greatest amount of ghrelin is secreted from gastric cells rather than from the hypothalamus. Plasma ghrelin concentrations increase when fasting, and decrease after food intake [7].
Mechanism of GH Action
GH stimulates linear body growth through differentiation and proliferation of the cells in the growth plate in children. GH also acts on many peripheral tissues other than the growth plate, and plays important roles in homeostasis, such as glycemic effects, hydration, protein anabolism, and lipid degradation [8] (Fig. 8.1).
At least part of the growth effect by GH is through endocrine, autocrine, and paracrine mechanisms of insulin-like growth factor I (IGF-I). GH action in body growth may be explained through three pathways involving IGF-I. In one pathway, GH acts through GH receptor (GHR) expression in hepatocytes and generation of IGF-I [9]. Consequently, serum IGF-I levels increase and IGF-I acts on peripheral tissues as a hormone by an endocrine mechanism. In a second pathway, GH acts on peripheral tissues, not the liver, promoting IGF-I generation, and this IGF-I affects local tissues by an autocrine/paracrine system [10]. Expression of GHR, IGF-I, and IGF-I receptor has been detected in chondrocytes, osteoblasts, osteoclasts, myocytes, and adipocytes. In a third pathway, GH affects peripheral tissues directly. For Laron syndrome in which there is deficiency of GHR, extrinsic IGF-I administration does not have a sufficient effect on growth in spite of its biological activities, such as improvement of hyperglycemia [11]. This phenomenon implies a direct action of GH.
GH Treatment
History
In the 1950s, human GH was first used to stimulate linear growth in a child with hypopituitarism [12]. At that time, GH was extracted and purified from the pituitary grand. Because the supply of the extracted GH was limited, GH treatment was restricted to children with the most severe and unequivocal GH deficiency (GHD). Delays in diagnosis and treatment, interruptions in treatments, and dosage restrictions were common during this time. Consequently, while GH accelerated growth of these individuals, adult height was usually less than average [13–15].
In 1985, Creutzfeldt–Jakob disease (CJD) was recognized in patients who had received GH. Distribution of pituitary-derived GH was stopped. Subsequently, in the United States, CJD was diagnosed in seven recipients of GH [16, 17]. Fortunately, 192- and 191-amino-acid biosynthetic GHs were approved in 1985 and 1987, respectively. The production of GH by biological systems transplanted with the GH gene yields a virtually unlimited supply of GH.
Biosynthetic GH treatment eliminated the risk of CJD and offered children with severe GHD an opportunity for optimal treatment. Children with milder forms of inadequate GH secretion, previously excluded from receiving GH, could become treated. In addition, metabolic effects of GH, apart from linear growth promotion, are now being studied extensively, leading to new indications for GH treatment [18].
Approved Disorders and the Efficacy of GH Treatment
Approved disorders for GH treatment have been expanding in the world in spite of its high cost, with expectations of promoting linear growth. Currently, these growth disorders are GHD, short children with small for gestational age (SGA), Turner syndrome (TS), chronic renal insufficiency (CRI), Prader–Willi syndrome (PWS), short stature homeobox (SHOX) haploinsufficiency, achondroplasia (ACH), hypochondroplasia (HCH), Noonan syndrome (NS), and idiopathic short stature (ISS) (Table 8.1). The type of approved disorders, criteria of diagnosis, and treatment dose vary and depend on the country.
Table 8.1
Approved diseases for GH in various countries as of 2013
GHD | Adult GHD | SGA | TS | CRI | PWS | SHOX haploinsufficiency | ACH | HCH | NS | ISS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
USA | ○ | ○ | ○ | ○ | – | ○ | ○ | – | – | ○ | ○ |
UK | ○ | ○ | ○ | ○ | ○ | – | ○ | – | – | – | – |
France | ○ | ○ | ○ | ○ | ○ | ○ | ○ | – | – | – | – |
Germany | ○ | ○ | ○ | ○ | ○ | ○ | ○ | – | – | – | – |
Sweden | ○ | ○ | ○ | ○ | ○ | ○ | – | – | – | – | – |
Japan | ○ | ○ | ○ | ○ | ○ | ○ | – | ○ | ○ | – | – |
Taiwan | ○ | ○ | ○ | ○ | – | – | – | – | – | – | – |
Australia | ○ | ○ | – | ○ | ○ | – | – | – | – | – | – |
GH treatment was started primarily for classical GHD patients to promote linear growth. Untreated patients with GHD have profound short stature, averaging nearly −5 standard deviation (SD) [19–21]. In many countries, pediatric endocrinologists have developed guidelines for diagnosis, criteria for starting treatment, treatment regimens, criteria for continuing treatment, and criteria for finishing treatment. GH treatment in GHD patients gradually improved their adult height SD score by approximately −1.3 SD, although most patients failed to reach their genetic target heights [22, 23].
SGA is a term used to describe a neonate’s birth size based upon appropriate auxological standards for healthy infants. Approximately 86 % of SGA children achieve a length within the normal range by 12 months [24, 25]. Catch-up growth in the normal range is virtually always complete by 2 years of age [26]. Overall, 8–14 % of SGA infants become short in stature with an adult height of approximately 1 SD [27, 28]. SGA children achieve a final height within the normal height range after 7.8 years of GH treatment [29]. The effects of GH extend beyond linear growth and potentially include important effects on body composition, muscle mass and function, bone mass, metabolism, behavior, and cognitive function, and even quality of life, IQ, and bone mineral content [30, 31].
TS is characterized by short stature, cubitus valgus, webbing of the neck, and sexual infantilism [32]. Over 95 % of TS patients eventually fall below the −2 SD, and their adult height is typically approximately 20 cm below the mean for females of their respective ethnic group. GH treatment in TS patients improves their final height to 8.5 cm above the mean projected adult height and there is a mean height gain due to GH of +7.2 cm [33, 34].
Growth failure is still a major obstacle to successful rehabilitation of children with CRI. The mean height SD score at the start of renal replacement therapy is approximately −2, indicating that half of the patients have a short stature [35, 36]. Similarly, the mean final height SD score of CRI patients is reported to be significantly reduced and varies between −1.4 in girls and −2.2 in boys in various reports [37, 38]. GH treatment for short stature in CRI became available approximately 20 years ago [39]. The final height of CRI patients after extended GH treatment appears to be an average of 1.0–1.5 SD [40, 41].
PWS is a neurogenetic disorder characterized by mental and physical abnormalities. The mean adult height achieved by men and women with PWS is 155–162 and 148–150 cm, respectively [42, 43]. The GH-deficient state commonly associated with PWS, as evidenced by reduced GH secretion, low serum IGF-I levels, and clinical features typical of GHD, has provided a rationale for trials assessing the efficacy of GH treatment. However, currently, the duration of treatment is limited. Longitudinal growth has been shown to increase by GH treatment in PWS [44, 45]. Some reports have shown that growth continues to improve by GH treatment in PWS, with the result that the target height SD score can be reached [46].
NS is a genetic syndrome with many features similar to TS, and is characterized by pulmonary valvular stenosis, visual problems, clotting disorders, and short stature [47]. Although approximately half of NS patients will reach an adult height within 2 SD of the population mean, the mean adult height of NS is approximately 162.5 cm and 153 cm for males and females, respectively [48]. GH treatment in NS improves the final height SD score to 1.7 [49]. Additionally, pretreatment baseline cortical bone mineral density (BMD) is reported to be in the low–normal range and it increases over 2 years of GH therapy [50]. In the majority of reports, GH treatment induced catch-up growth in most of the NS patients. First data on long-term outcome demonstrate an effect comparable with or even better than that in TS.
ISS is a purely descriptive term that refers to a child, adolescent, or adult with a height below the age reference for population and sex, in whom, with current diagnostic tools, no etiological diagnosis is made [51]. The mean final height is similar to the mean predicted height in ISS. There is a large interindividual variation that is primarily correlated with the initial height SD score and bone age delay at start of GH treatment [52]. GH for ISS in a supraphysiological dosage increases the final height by approximately 7 cm, but for the individual child, the height gain is difficult to predict.
Side Effects
Recombinant biosynthetic GH preparations are highly purified and free of contaminants. The possibility of viral transmission through GH has been virtually eliminated. Antigenicity of GH preparations is also low, although GH antibodies can be detected in 10–30 % of treated children [53]. With rare exceptions (less than 0.1 %), these antibodies do not impede effects of GH.
Laboratory indications of hypothyroidism can be found in as many as 25 % of GHD children treated with GH [54]. GHD patients, who display subnormal nocturnal thyroid-stimulating hormone surges, signifying preexisting central hypothyroidism, are more likely to display subnormal T4 and free T4 levels during GH therapy [55]. However, most studies have indicated that children with normal thyroid function before treatment do not develop significant perturbations in thyroid hormone metabolism during GH therapy.
Administration of unphysiological high concentrations of GH may lead to defects in glucose metabolism [56]. When intrinsic GH secretion is increased, as in sleep, oral or intravenous glucose tolerance tests show a defect in glucose metabolism [57]. This defect of glucose metabolism lasts even after finishing GH treatment and normalization of serum GH concentrations.
Edema and sodium retention rarely occur early in the course of GH therapy, which is attributable to an anti-natriuretic effect on the renal tubules of GH and/or IGF-I. Minor elevations in plasma renin activity and aldosterone observed in the first 3 days of treatment resolve within 1 or 2 weeks [58]. Occasionally, fluid shifts within the central nervous system are sufficient to cause benign intracranial hypertension, with symptoms of headache, visual loss, vomiting, and papilledema. Direct fluid-retaining properties of GH and/or action of locally produced IGF-I on cerebrospinal fluid production are speculated to be causative. Cessation of GH therapy reverses symptoms in spite of continued GH treatment [59]. Resumption of GH treatment has been successfully accomplished with re-initiation at a lower dosage and a gradual return to the initial dosage. Performing a fundoscopic examination is recommended in all patients before initiation of GH therapy and periodically thereafter [60].
Growth Hormone and Bone
Effect of GH on Bone and Cartilage Metabolism
GH acts directly on the perichondrial layer in the growth plate of growing bones, and promotes proliferation and differentiation of pre-chondrocytes, as well as promotes IGF-I synthesis (Fig. 8.2). Pre-chondrocytes proliferate and differentiate to chondrocytes in the proliferative zone of growth plates by acquiring the ability for reaction to IGF-I and for generation of IGF-I [61].
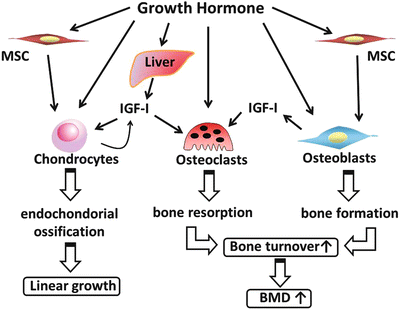
Fig. 8.2
Schematic representation of bone metabolism by GH. GH stimulates differentiation and proliferation of chondrocytes directly and through IGF-I synthesis. This endochondral ossification leads to linear growth. GH also stimulates differentiation and proliferation in osteoblasts and osteoclasts. Consequently, GH affects bone metabolism and, consequently, linear growth and bone mineral density
Osteoblasts express GHR and IGF-I receptor, and have the ability of generating IGF-I [62]. Therefore, GH promotes synthesis of IGF-I in osteoblasts, and IGF-I acts on these cells through the autocrine/paracrine system. In osteoblastic culture, IGF-I stimulates differentiation of osteoblasts to osteocytes by promoting proliferation of osteoblasts, expression of type I collagen, and activation of alkaline phosphatase, and by suppressing expression of matrix metalloproteinase 1 [63]. However, it is still unclear whether GH action on osteoblasts occurs through IGF-I or there is a direct pathway.
GH action is also detected in osteoclasts. Precursors of osteoclasts express GHR, and GH promotes their differentiation to osteoclasts. Factors promoting osteoclast differentiation are generated by GH-stimulated osteoblasts and bone marrow cells [64]. These findings show that GH promotes bone resorption through its direct effect on bone marrow cells or through osteoblasts. In fact, when GH is administered in pediatric patients, bone resorption markers are elevated before growth is detected and bone formation markers are elevated.
GH promotes bone turnover, including bone generation and bone resorption; GH consequently promotes longitudinal bone growth while maintaining BMD suitable for increasing quantities of bone (Fig. 8.2). Since GH also increases mass and strength of the skeletal muscles, mechanical stress may be another factor for GH effect on increasing BMD [65]. Although the effect of GH on BMD is still controversial in certain conditions such as burn injury, BMD is indeed correlated with nocturnal GH secretion in young healthy men and acromegaly [66, 67]. Moreover lumbar BMD is reduced in pediatric GHD patients, and GH treatment increases BMD in GHD and other diseases [68–71].
For the considerable variability in response to GH treatment, several prediction models that attempt to estimate the growth response to GH treatment have been developed [72–75]. As a result, in growing children, markers of bone metabolism reflect skeletal growth and development. For example, urinary deoxypyridinoline and serum pyridinoline, bone resorption markers, are strongly related to height velocity. These results imply that bone metabolism and linear growth are closely related to each other.
Approved GH Treatment in Skeletal Dysplasia
Skeletal dysplasia is a heterogeneous group of diseases affecting the skeleton. The estimated incidence is 30–45 in every 100,000 newborns. The final height differs substantially between the various disorders, but is often in the range of 110–130 cm [76]. Currently, although a remarkably short stature has been detected in various skeletal dysplasias, only three skeletal dysplasias have been approved for GH treatment: ACH, HCH and SHOX haploinsufficiency [77] (Table 8.1).
ACH is the most common type of rhizomelic short-limb dwarfism caused by activating point mutations in the fibroblast growth factor receptor 3 (FGFR3) gene [78, 79]. The incidence of ACH is estimated as 1 in 25,000 live births. The average adult height of ACH is approximately 132 cm (−6.8 SD) for males and 124 cm (−6.4 SD) for females [80]. FGFR3 is expressed in the growth plate, and its activation suppresses IGF-I expression and cell proliferation, and promotes apoptosis of chondrocytes. GH administration increases IGF-I expression in chondrocytic cell lines expressing mutated FGFR3 and prevents these cells from apoptosis [81]. This could explain one of the mechanisms by which GH therapy improves disturbed bone growth in ACH.
GH treatment in ACH has been approved only in Japan, since 1997. As a short-term effect, GH administration increases height velocity from (mean ± SD) 3.8 ± 0.9 to 6.6 ± 1.6 cm/year in patients with ACH for at least 6 months [81]. In longer-term studies, GH treatment in ACH patients promotes their height velocity in the first treatment year and promotes their linear growth, with a gain of 1–1.5 SD over 3–6 years, although height velocity is low after the second year of treatment (Fig. 8.3) [82–84]. More than 15 years have passed since approval, but reports on the long-term effect of GH on ACH regarding the prognosis of height and bone mineral metabolism have still not been published.
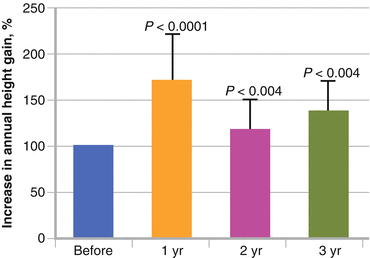
Fig. 8.3
Short-term effect of GH treatment in achondroplasia (ACH). The graph shows the percentage increase in height in ACH patients. Growth velocity is increased when GH treatment is started and is maintained at a higher level than that before GH treatment during 3 years [83]
HCH is also mainly caused by mutations of the FGFR3 gene and is characterized by short stature and abnormal body proportions, although not as severe as in ACH. The final height in HCH is compromised and in the range of 132–147 cm [85, 86]. GH treatment for HCH has been approved only in Japan at the same time as ACH in 1997. Several reports have shown that the median height SD score is approximately −3.2 SD at the start of GH therapy for HCH and it improves plus 1 SD after 2–5 years of GH treatment [87, 88]. Because some HCH patients have no mutation in the FGFR3 gene, but characteristic facial features, bone deformities, and disproportionate short stature are observed, there are still some doubts as to the certainty of the diagnosis in some of the patients diagnosed with HCH. Therefore, clinical studies of GH treatment, including genetic background data, are required.
Dyschondrosteosis, or Leri–Weill syndrome, is a mesomelic skeletal disorder caused by a deletion or mutation in the SHOX gene [89]. In dyschondrosteosis, there are abnormal proportions due to short legs, and the adult height in these individuals is variable, but in most patients it is reduced. However, a reduction in height appears to be sex-specific, with a greater loss of height in females compared with males [90]. Isolated SHOX haploinsufficiency is observed in 56–100 % of patients with Leri–Weill dyschondrosteosis and in 1–14 % of ISS [91]. Short stature observed in patients with TS is partially explained by haploinsufficiency of the SHOX gene [92]. Because GH treatment in TS improves the final height SD score, GH treatment in patients with SHOX haploinsufficiency has been approved in some countries. Prepubertal children with isolated SHOX defects treated with GH during 2 years present with a similar growth response to that of TS patients [93] and reach their final height with a height SD score gain of 1.1 ± 0.7 after 4.7 years [94]. The gain in the height SD score during the first year of GH therapy for patients with SHOX haploinsufficiency shows an increase of 0.7 SD [95]. The sitting height ratio SD score does not change during 1 year of GH treatment in patients with SHOX haploinsufficiency. Adult height in GH treatment for dyschondrosteosis has not been published yet.
Challenging Trials of GH Treatment
GH treatment has been attempted in many diseases with short stature, such as Down syndrome, Cornelia de Lange syndrome, Kabuki syndrome, Fanconi anemia, Rubinstein–Taybi syndrome, Klippel–Feil syndrome, Diamond–Blackfan anemia, and skeletal dysplasia. We discuss below regarding GH treatment in skeletal dysplasia, focusing on GH and bone, such as osteogenesis imperfecta (OI) and X-linked hypophosphatemic (XLH) rickets. Because the final height of each disorder has not been determined yet, further evidence of GH treatment in all challenging disorders needs to be gathered.
Osteogenesis Imperfecta
OI is an autosomal dominant disorder caused by dysfunction of type I collagen proteins. OI is characterized by congenital-decreased BMD, bone fragility, short stature, blue sclerae, progressive bone deformities, and dentinogenesis imperfecta [96, 97]. Clinical severity varies widely from lethal to mild with non-deformity. Recently, OI patients were classified into eight types according to their severity [98, 99].
The most popular internal treatment of OI is bisphosphonates suppressing bone resorption. Bisphosphonates in OI children increase BMD and result in dramatically decreased bone fractures [97]. Because growth deficiency is constantly present in severe OI and common in mild to moderate forms of OI, GH could be used in OI for stimulating bone metabolism or for increasing linear growth [100, 101].
Although there are few reports of GH treatment in patients with OI, GH action positively affects bone growth and bone turnover by stimulating osteoblasts, collagen synthesis, and longitudinal bone growth [102, 103]. A recent study also suggested that combined bisphosphonate and GH treatment in OI patients for 1 year positively increases BMD and growth velocity, and does not affect the peripheral fracture rate [104]. Although GH treatment in OI has not been approved yet, GH may be expected to improve symptoms of OI patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


