62 Glomerulonephritis
Etiology and Pathogenesis
Most glomerulonephritides result from either circulating immune complex deposition within the glomerulus or in situ immune complex formation. This activates complement, as well as cellular and humoral pathways of inflammation. Within the glomerulus, there can be endothelial, epithelial, and mesangial hypercellularity, infiltration of leukocytes, thickening or duplication of the glomerular basement membrane, and necrosis (Figure 62-1). This results in loss of capillary integrity and obstruction of blood flow through the glomerular capillary loops. This capillary injury and obstruction of glomerular blood flow leads to the fluid overload, oligoanuric renal failure, hematuria, and RBC casts.
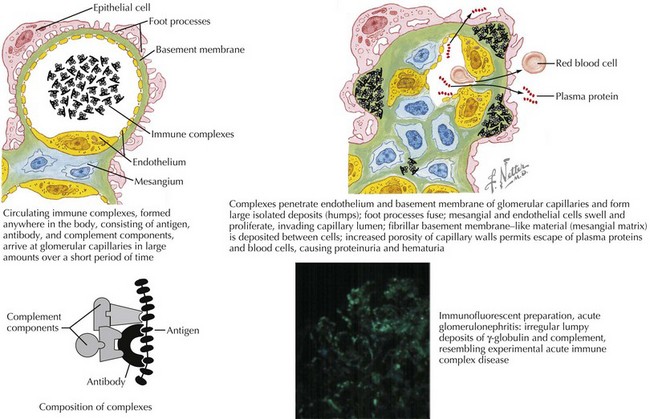
Differential Diagnosis
Acute Postinfectious Glomerulonephritis
Acute poststreptococcal GN (APSGN) is the most common cause of APIGN in children and has a higher incidence in developing countries (Figure 62-2). APSGN is caused by nephritogenic forms of Lancefield group A streptococci. In most cases, there is clinical or laboratory evidence of antecedent streptococcal infection. APSGN must be confirmed or ruled out before looking for less common causes of post infectious GN (Staphylococcus aureus, Streptococcus viridans).
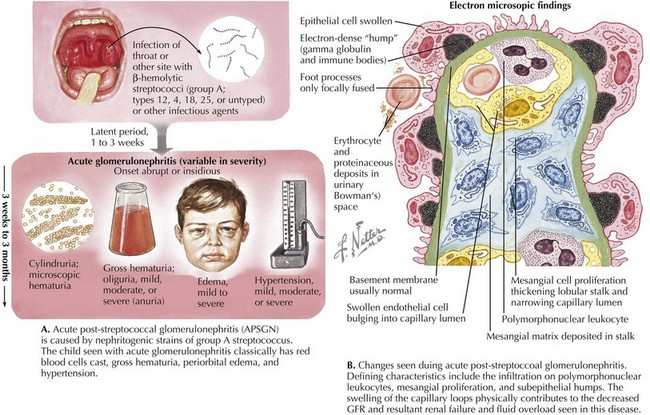
By light microscopy, findings in APSGN include glomerular enlargement, mesangial cell expansion and proliferation, and neutrophil exudation (see Figure 62-2). Electron microscopy (EM) reveals discrete subepithelial deposits.
Henoch-Schönlein Purpura Nephritis
HSP nephritis is a small vessel vasculitis caused by IgA deposition within the glomeruli in the context of systemic HSP. Renal manifestations may present weeks after the onset of systemic HSP; rarely is it the first feature manifested in this syndrome. Prevalence of renal manifestations is subject to observer bias. Pediatric nephrology centers report that 50% of children with HSPN have hematuria and proteinuria, 8% have acute GN (AGN), 13% have nephrotic syndrome, and 29% have a mixed nephritic and nephrotic syndrome. Treatment of HSP nephritis is controversial because of a high rate of spontaneous remission and the lack of rigorous studies regarding treatment. Prognostic features are noted in Table 62-1.
Table 62-1 Poor Prognostic Features of Selected Glomerulonephritides and Recommended Treatment
| Disease | Poor Prognostic Features | First-Line Treatment |
|---|---|---|
| IgAN | Proteinuria >1 g/24 h, hypertension, azotemia, interstitial fibrosis, sclerotic glomeruli | If medical treatment,- corticosteroid ACEI |
| HSP | Presence of nephritic or nephrotic syndrome, renal failure, nephrotic-range ongoing proteinuria, interstitial fibrosis, sclerotic glomeruli | If medical treatment needed, corticosteroids, immunosuppressive agents |
| SLE | Diffuse proliferative GN (WHO Class IV), ↑ creatinine, persistent HTN, chronic anemia, nephrotic-range proteinuria | Corticosteroid therapy, cyclophosphamide, azathioprine, MMF |
| ANCA+ vasculitis or pauci-immune GN | Crescents on biopsy, frequent relapses | Corticosteroid therapy, azathioprine, MMF, cyclophosphamide |
| MPGN | Type II disease, nephrotic-range proteinuria | Corticosteroid therapy |
ANCA, antineutrophilic cytoplasmic antibody; AZA, azathioprine; CCS, corticosteroids; GN, glomerulonephritis; HSP, Henoch-Schönlein purpura; HTN, hypertension; IgAN, IgA nephropathy; MMF, mycophenolate mofetil; MPGN, membranoproliferative glomerulonephritis; SLE, systemic lupus erythematosus; WHO, World Health Organization.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


