CHAPTER 43 Gestational trophoblastic tumours
Introduction
The World Health Organization (WHO) has classified gestational trophoblastic disease (GTD) into two premalignant diseases, termed ‘complete hydatidiform mole’ (CM) and ‘partial hydatidiform mole’ (PM), and three malignant disorders, termed ‘invasive mole’, ‘gestational choriocarcinoma’ and ‘placental-site trophoblastic tumour’ (PSTT) (World Health Organization 1983). These malignant disorders are also frequently referred to as ‘gestational trophoblastic tumours’ (GTTs) or ‘neoplasia’ (GTN). GTTs are important to recognize because they are nearly always curable and fertility can be preserved in most cases. This is mainly because:
Genetics and Pathology
Complete hydatidiform mole
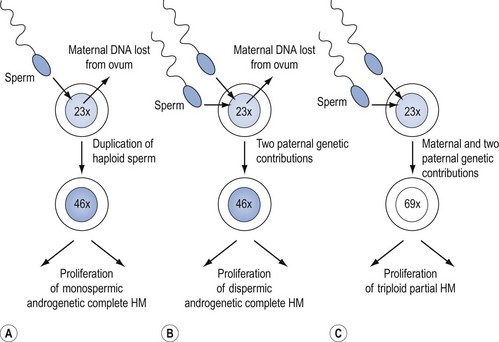
CMs nearly always contain paternal DNA alone and are therefore androgenetic. This occurs in most cases because a single sperm bearing a 23X set of chromosomes fertilizes an ovum lacking maternal genes, and then duplicates to form the homozygote, 46XX (Figure 43.1A). However, in up to 25% of CMs, fertilization can take place with two spermatozoa, resulting in the heterozygous 46XY or 46XX configuration (Figure 43.1B). A 46YY conceptus has not yet been described and is presumably non-viable. Very rarely, a CM can arise from a fertilized ovum which has retained its maternal nuclear DNA and is therefore biparental in origin (Fisher and Newlands 1998). Macroscopically, the classic CM resembles a bunch of grapes due to generalized (complete) swelling of chorionic villi. However, this appearance is only seen in the second trimester and the diagnosis is usually made earlier when the villi are much less hydropic. Indeed, in the first trimester, the villi microscopically contain little fluid, are branching and consist of hyperplastic syncytio- and cytotrophoblast with many vessels. Although it was previously thought that CM produced no fetal tissue, histology from 6–8-week abortions reveals evidence of embryonic elements, including fetal red cells (Paradinas 1998). This has resulted in pathologists incorrectly labelling CMs as PMs. The presence of embryonic tissue from a twin pregnancy comprising a fetus and a CM is another source of error which can lead to the incorrect diagnosis of PM.
Partial hydatidiform mole
Genetically, PMs are nearly all triploid with two paternal and one maternal sets of chromosomes (Figure 43.1C). Rarely, tetraploid PMs are found. Triploidy occurs in 1–3% of all recognized conceptions and in approximately 20% of spontaneous abortions with an abnormal karyotype, but at least two paternal sets of chromosomes are needed for PM development. Triploids due to two sets of maternal chromosome do not become PMs (Lawler et al 1982). Although a variety of reports have suggested that diploid PMs exist, genetic analysis of such lesions has not supported this. In general, a diploid molar gestation is believed to be a CM (Genest et al 2002). Flow cytometry, which can be done in formalin-fixed, paraffin-embedded tissues (Seckl et al 2000), can help in differentiating CM from PM, and PM from diploid non-molar hydropic abortions.
In PMs, villous swelling is less intense and only affects some villi. Both swollen and non-swollen villi can have trophoblastic hyperplasia which is mild and focal. The villi have characteristic indented outlines and round inclusions. An embryo is usually present and can be recognized macroscopically or inferred from the presence of nucleated red cells in villous vasculature. It may survive into the second trimester, but usually dies at approximately 8–9 weeks of gestation and this is followed by loss of vessels and stromal fibrosis. In PMs evacuated early, villous swelling and trophoblastic excess can be so mild and focal that the diagnosis of PM may be missed (Paradinas 1998). Indeed, at uterine evacuation for a ‘miscarriage’, it is likely that many PMs are misclassified as products of conception. Fortunately, the authors only see about one patient per year with persistent GTD related to a previously unrecognized PM. Of the increasing number of PMs which are correctly diagnosed, very few go on to develop persistent GTD. Indeed, in approximately 3000 PMs reviewed and followed at Charing Cross Hospital between 1973 and 1997, only 15 (0.5%) have required chemotherapy (Seckl et al 2000).
Other pregnancies mistaken for hydatidiform mole
Over half of first-trimester non-molar abortions are due to trisomy, monosomy, maternally derived triploidy and translocations. These often develop hydrops but are small (<3 mm), and PM can be excluded if they are diploid on flow cytometry. A significant recent development in the pathological analysis of hydatidiform mole (HM) is the use of p57kip2 immunostaining to make a definitive diagnosis of androgenetic CM as opposed to a hydropic abortion or a PM. p57kip2, a cyclin-dependent kinase inhibitor, is a paternally imprinted gene which is maternally expressed. The absence of maternal genes in androgenetic CM means that the gene cannot be expressed in CM cytotrophoblast. Consequently, p57kip2 staining is negative with CMs, in contrast to PMs, hydropic abortion and normal placenta. This technique is well validated, easy and inexpensive to perform (Sebire et al 2004, Popiolek et al 2006).
Syndromes such as Turner’s, Edward’s and Beckwith-Wiedemann’s can also cause histological confusion with PMs (Paradinas 1998).
Invasive hydatidiform mole
Invasive hydatidiform mole is common and is clinically identified by the combination of an abnormal uterine ultrasound (US) and a persistent or rising hCG level following uterine evacuation of a CM or PM. Pathological confirmation of this condition is rarely required. Moreover, repeat dilatation and curettage (D & C) is often contraindicated because of the risks of uterine perforation, infection, life-threatening haemorrhage and subsequent hysterectomy. In occasional cases where histology is available, invasive mole can be distinguished from choriocarcinoma by the presence of chorionic villi. Identification of cases of CM and PM that will subsequently undergo malignant transformation would be an important development. There is accumulating evidence that such HMs are more likely to show telomerase activity (i.e. have limitless replicative potential) and increased expression of the anti-apoptotic gene Mcl-1 (Wells 2007). These findings need further prospective evaluation.
Choriocarcinoma
Most choriocarcinomas have been shown to have grossly abnormal karyotypes with diverse ploidies and several chromosome rearrangements, none of which are specific for the disease (Arima et al 1994). Studies of the origin of GTTs have confirmed that choriocarcinoma may arise from any type of pregnancy including a normal term pregnancy, or homozygous or heterozygous CM. Until recently, it was thought that PMs could not give rise to choriocarcinoma. However, there is now incontrovertible genetic evidence that PMs can indeed transform into choriocarcinomas (Seckl et al 2000). This is important as some centres wrongly believe that it is safe to discontinue hCG follow-up following the diagnosis of a PM.
Interestingly, choriocarcinoma may not always be due to the antecedent pregnancy. A patient with a history of CM 4 years previously developed choriocarcinoma following the delivery of a twin pregnancy. Using polymerase chain reaction to amplify short tandem repeat polymorphisms in DNA, this tumour was shown to be genetically identical to the previous CM (Fisher et al 1995). As for invasive moles, obtaining tissue to make a formal histological diagnosis of choriocarcinoma is often not appropriate, so doubt frequently exists regarding whether patients have one or the other form of GTT.
Placental-site trophoblastic tumour
PSTTs have been shown to follow term delivery, non-molar abortion or CM. Furthermore, a recent case has shown that PSTTs can develop after a PM (Palmieri et al 2005). Like choriocarcinoma, the causative pregnancy may not be the immediate antecedent pregnancy (Fisher et al 1995). Genetic analysis of some PSTTs has demonstrated that they are mostly diploid, originating from either a normal conceptus and therefore biparental, or androgenetic and arising from a CM (Newlands et al 1998a).
In the normal placenta, placental-site trophoblast is distinct from villous trophoblast and infiltrates the decidua, myometrium and spiral arteries of the uterine wall. PSTTs are rare, slow-growing malignant tumours composed mainly of intermediate trophoblast derived from cytotrophoblast, and so produce little hCG. However, they often stain strongly for human placental lactogen (hPL) and β1-glycoprotein. Elevated Ki-67 levels may help in distinguishing PSTTs from regressing placental nodules (Shih and Kurman 1998). In contrast to other forms of GTT, spread tends to occur late by local infiltration and via the lymphatics, although distant metastases can occur. More than five mitoses per 10 high-power fields may predict tumours with metastasizing potential (Newlands et al 1998a).
Epithelioid trophoblastic tumour
Epithelioid trophoblastic tumours (ETTs) are a recently described neoplastic proliferation of intermediate trophoblast that are thought, by some investigators, to be distinct from PSTTs and choriocarcinoma. It has been proposed that ETTs arise from the intermediate trophoblasts of the chorionic laeve (Shih and Kurman 2001). Histologically, ETTs display a relatively uniform, nodular proliferation of intermediate-sized epithelioid trophoblasts, forming nests and cords. Islands of trophoblast are typically surrounded by areas of hyalinization or eosinophilic debris simulating tumour cell necrosis and resembling keratinous material in a squamous cell carcinoma. ETTs can be associated with focal replacement of the cervical glandular epithelium with stratified neoplastic cells, simulating squamous cervical intraepithelial neoplasia. ETT cells are positive for cytokeratin, epithelial membrane antigen and inhibin-A, whereas trophoblastic markers hPL, hCG and melanoma cell adhesion molecular are only expressed focally (Hui et al 2005).
Epidemiology and Aetiological Factors
Hydatidiform mole
Incidence and ethnic origin
The incidence of HM in the UK is one in 714 pregnancies (Tham et al 2003). Recent results indicate that the previously documented higher rates in the Far East have fallen towards the stable levels found in Europe and North America (Hando et al 1998), possibly because of dietary changes. The incidence of PM has been underestimated in the past and is currently three per 1000 pregnancies (Newlands et al 1998b).
Age
CMs are more common at the extremes of reproductive age. In one study, compared with the lowest rate between 25–29 years, the relative increased risk was six-fold in girls under 15 years, three-fold between 40 and 45 years, 26-fold between 45 and 49 years and more than 400-fold over 50 years of age (Bagshawe and Begent 1983, Newlands et al 1998b). PMs are also more common at the extremes of reproductive age, although the effect is less pronounced (Sebire et al 2002a).
Previous pregnancies
Increasing gravidity does not increase the risk of CM. However, following one CM, the risk of a subsequent pregnancy being a CM rises from one in 1000 to one in 76, and to one in 6.5 with two previous CMs (Bagshawe and Begent 1983). Therefore, patients with a previous CM must be followed-up after each subsequent pregnancy to confirm that their hCG levels return to normal. Similar results have been reported for PMs, with the risk for subsequent molar pregnancies rising to one in 59 for women with one previous PM (Sebire et al 2003).
Choriocarcinoma
The incidence of choriocarcinoma following term delivery without a history of CM is approximately one in 50,000. However, CM is probably the most common antecedent to choriocarcinoma, comprising 29–83% in various studies across the world (World Health Organization 1983). Consequently, the overall incidence of choriocarcinoma after a CM is much higher. Proof of this is frequently difficult to obtain, but when histology was available, these tumours were identified as choriocarcinoma in 3% and invasive mole in 16% of previous CMs (seldom as PSTT). Rarely, PMs can give rise to choriocarcinoma (Seckl et al 2000). Unlike HM, choriocarcinoma does not exhibit any clear geographical trends in incidence, but the effect of age remains important.
Placental-site trophoblastic tumours
First described as a separate disease entity in 1976 (Kurman et al 1976), there are currently approximately 150 recorded cases of this tumour in the literature; therefore, estimates of its true incidence may be quite inaccurate (Newlands et al 1998a, Papadopoulos et al 2002, Hassadia et al 2005, Baergen et al 2006). In the largest series described — 62 patients between 1976 and 2006 — PSTTs represented 0.2% of all trophoblastic tumours (Schmid et al 2008) The relative incidence in the UK has been stable over the past 15 years following an increase in the first years after the introduction of this new disease classification.
Genetic Factors: The Role of Imprinting
Hydatidiform mole
All autosomal genes consist of two alleles (paternal and maternal). However, some alleles are only expressed from one parent and not the other; a phenomenon called ‘genomic imprinting’. Underexpression of an imprinted gene has been demonstrated in cases of both familial and sporadic CM, and abnormal methylation patterns have been described in another family (Fisher et al 2002, Judson et al 2002).
Extensive mapping studies have been performed on the families with CM. These have demonstrated a defective locus at 19q13.4 in five families which has been localized to a single gene — NALP7 (Murdoch et al 2006). This is the first causative single gene defect identified in CM. NALP7 is a member of the CATERPILLER family which is involved in inflammation and apoptosis. It is expressed in oocytes and the endometrium, and is a negative regulator of the proinflammatory cytokine interleukin-1β, which is involved in regulating trophoblast invasion during implantation. It is unclear how defects in NALP7 result in CM formation, but abnormal inflammation during embryogenesis may be involved.
NALP7 does not appear to be involved in the establishment of imprinting; therefore, defects in NALP7 may be a consequence of abnormal imprinting rather than its cause. Further families have been identified that do not map to 19q13.4, proving that familial CM is a genetically heterogeneous condition (Zhao et al 2006). Moreover, to date, abnormal NALP7 has not been demonstrated in sporadic CM or PM cases.
Three other, closely related, genes are imprinted and may be involved in GTT development. These are H19, a putative tumour suppressor gene (Hao et al 1993), and p57kip2 (discussed previously) (Matsuoka et al 1996), both of which are normally expressed by the maternal allele; and the paternally expressed IGF-2, a growth factor commonly implicated in tumour proliferation (Ogawa et al 1993). While p57kip2 showed the expected pattern of expression in CM and choriocarcinoma (Chilosi et al 1998), CM and postmole tumours were unexpectedly found to express H19 (Walsh et al 1995), and some post-term tumours showed biallelic expression of both H19 and IGF-2 (Hashimoto et al 1995). This suggests that loss of the normal imprinting patterns of these genes may be an important factor in the development of GTT.
Choriocarcinoma
Historically, cytogenetic studies of choriocarcinoma have demonstrated a diverse range of abnormalities. Microsatellite studies have shown loss of heterozygosity at specific regions: deletions of 7p12-q11.2, amplification of 7q21-q31 and loss of 8p12-p21 (Matsuda et al 1997) Abnormalities in the latter two are also found in some cases of ovarian and breast cancer, and are postulated to encode novel tumour suppressor genes. Several groups have reproduced these findings in non-molar choriocarcinoma, but results in postmolar choriocarcinoma have been inconsistent (Burke et al 2006). Only a minority of tumours showed loss of heterozygosity for all three regions, suggesting that these defects are acquired late in the development of choriocarcinoma and are not essential for malignant transformation.
Risk of Gestational Trophoblastic Tumours Following Complete or Partial Hydatidiform Mole
Following evacuation of a CM or PM, the risk of developing a GTT is less than 16% and 0.5%, respectively (Bagshawe et al 1990, Seckl et al 2000). Since it is not yet possible to predict which patients with a CM or PM will develop persistent GTD, all patients must be registered for hCG monitoring. Following this strict protocol enables the identification of individuals with persistent trophoblastic growth who could benefit from life-saving chemotherapy.
Human Chorionic Gonadotrophin
Beta-human chorionic gonadotrophin assays
The family of pituitary/placental glycoprotein hormones includes hCG, follicle-stimulating hormone, luteinizing hormone (LH) and thyroid-stimulating hormone (TSH). Each hormone comprises an α-subunit which is common between the family members and a distinct β-subunit. Consequently, assays to measure hCG are directed against the β-subunit. Many different β-hCG assays are available. Some detect intact β-hCG, and others are either selective for individual fragments or detect various combinations of fragments (Cole 1998). In pregnancy, hCG is usually intact and fragments of β-hCG are not produced, although the β chain is hyperglycosylated during the first trimester. However, in cancer, β-hCG may circulate in many different forms which can vary in their glycosylation status. Therefore, assays used in patients with GTD and cancer need to be able to detect all forms of β-hCG. The ideal assay should be able to recognize all forms equally well and be sufficiently sensitive to limit the risk of false-negative results (Mitchell and Seckl 2007). Moreover, the assay should not produce false-positive results as this is well recognized to be associated with unnecessary medical interventions and potentially life-threatening complications (Cole et al 2001). So how good are the existing β-hCG assays?
Commercially available β-hCG tests are based on the sandwich assay principle and rely on two antibodies which generally target different regions of the molecule. These assays are primarily licensed for use in pregnancy detection. However, they are frequently employed for monitoring patients with cancer. The assays can produce both false-positive and false-negative results (Cole et al 2001, Mitchell and Seckl 2007). The false-positive results occur because there is another molecule (often a heterophile antibody) which sticks the capture and detection antibodies together. This can usually be avoided by measuring the hCG in urine, as cross-reacting antibodies are large and cannot pass through the renal glomerulus into the urine. Alternatively, if a false-positive result is suspected, remeasuring the hCG on an alternative assay or serially diluting the serum sample usually resolves the issue. Real hCG will be seen in another assay and will dilute appropriately, whilst a cross-reacting molecule will be negative in another assay and does not serially dilute away. Recent work from the authors’ laboratory shows that commercial assays may be particularly prone to false-negative results, either because they completely fail to detect or as a consequence of poor sensitivity for a particular β-hCG isoform (Mitchell and Seckl 2007). False-negative results can potentially result in failure to diagnose disease or early termination of treatment, and therefore higher relapse rates.
In addition to the commercial assays, there are also several in-house assays in various centres around the world which are usually based on a single antibody to capture the hormone on a competitive basis with labelled (often radioactively) β-hCG. These assays may also produce false-positive and false-negative results. Indeed, all types of assay are only as good as the antibodies used. At Charing Cross Hospital, a rabbit polyclonal antibody is used in a radioimmunoassay (RIA); this recognizes all forms of β-hCG equally well and with sufficient sensitivity that false-negative results appear to be rare (Mitchell and Seckl 2007). Moreover, since this RIA is performed on both serum and urine samples which are serially diluted, the risk of false-positive results is very low (Mitchell and Seckl 2007).
New assays designed to detect specific hCG variants are in development and are leading to further increases in the specificity and sensitivity of hCG monitoring. Elevation of hyperglycosylated hCG (hCG-H) levels may be specific to cases of HM that subsequently require chemotherapy. This rise occurs earlier than in conventional hCG and before clinically apparent GTTs develop (Cole et al 2006a). However, further work is required, and since no commercial hCG-H assays are available, progress in this area is likely to be slow.
Recent data have also shown that proportionately higher levels of free β-hCG fragments are produced in PSTTs, and that this is a highly sensitive test for discriminating PSTTs from choriocarcinoma (Cole et al 2006b, Harvey et al 2008), although β-hCG may also be elevated in some non-trophoblastic malignancies.
Beta-human chorionic gonadotrophin as a tumour marker
hCG has a half-life of 24–36 h and is the most sensitive and specific marker for trophoblastic tissue. However, hCG production is not confined to pregnancy and GTD. Indeed, hCG is produced by any trophoblastic tissue found, for example, in germ cell tumours and in up to 15% of epithelial malignancies (Vaitukaitis 1979). The hCG levels in such cases can be just as high as those seen in GTD or in pregnancy. Therefore, measurements of hCG do not reliably discriminate between pregnancy, GTD or non-gestational trophoblastic tumours.
However, serial measurements of hCG have revolutionized the management of GTD for several reasons. The amount of hCG produced correlates with tumour volume, such that a serum hCG level of 5 IU/l corresponds to approximately 104–105 viable tumour cells. Consequently, these assays are several orders of magnitude more sensitive than the best imaging modalities available today. In addition, hCG levels can be used to determine prognosis (Bagshawe 1976). Serial measurements allow monitoring of disease progression or response to therapy (Figure 43.2). Development of drug resistance can be detected at an early stage, which facilitates appropriate changes in management. Estimates may be made of the time for which chemotherapy should be continued after hCG levels are undetectable in serum in order to reduce the tumour volume to zero. For these reasons, hCG is the best tumour marker known.
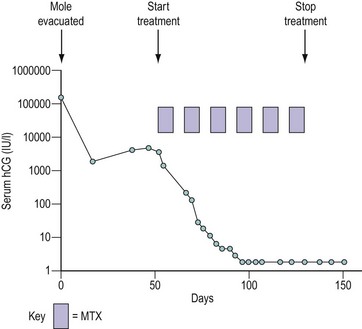
Figure 43.2 Graph demonstrating the use of monitoring the serum human chorionic gonadotrophin (hCG) concentration following evacuation of a hydatidiform mole (HM). In this case, after an initial fall, the hCG level started to rise, indicating the development of invasive HM or choriocarcinoma, so the patient was called up for staging. The prognostic score was low risk (see Table 43.4) and the patient was successfully treated with methotrexate (MTX) and folinic acid (see Table 43.5).
Clinical Features
Complete and partial moles
These most commonly present in the first trimester as a threatened abortion with vaginal bleeding. If the diagnosis is delayed, patients may notice the passing of grape-like structures (vesicles), and occasionally the entire mole may be evacuated spontaneously. The uterus may be any size but is commonly large for gestational age. Patients with marked trophoblastic growth and high hCG levels are particularly prone to hyperemesis, toxaemia and the development of theca lutein cysts which may sometimes be palpable above the pelvis. Toxaemia was diagnosed in 27% of patients with CM (Berkowitz et al 1981), but is seen less frequently today because of early US diagnosis. Convulsions are rare. The high hCG levels may also produce hyperthyroidism because of cross-reactivity between hCG and TSH at the TSH receptor. Although pulmonary, vaginal and cervical metastases can occur, they may disappear spontaneously following removal of the mole. Thus the presence of metastases does not necessarily imply that an invasive mole or choriocarcinoma has developed. Patients may rarely present with acute respiratory distress, not only because of pulmonary metastases or anaemia but occasionally as a result of tumour embolization (Savage et al 1998). The risk of embolization is reduced by avoiding agents which induce uterine contraction before the cervix has been dilated to enable evacuation of the CM.
Patients with PMs do not usually exhibit the dramatic clinical features characteristic of CM (Goldstein and Berkowitz 1994). The uterus is often not enlarged for gestational age, and vaginal bleeding tends to occur later so that patients most often present in the late first or early second trimester with a missed or incomplete abortion. In fact, the diagnosis is often only suspected when the histology of curettings is available. The pre-evacuation hCG level is less than 100,000 IU/l at diagnosis in over 90% of cases.
At present, a proportion of CMs and PMs still go undiagnosed because of miscarriage at home or because termination centres do not carry out histopathological examination of all abortions (Seckl et al 2004a). This can result in late presentation of disease, sometimes with life-threatening complications. Clearly, there is little that can be done about miscarriages at home. However, for women attending termination centres, it may be possible to establish a screening procedure to help prevent subsequent problems from missed diagnosis.
Twin pregnancies
Twin pregnancies comprising a normal fetus and an HM occur in between one in 20,000 and one in 100,000 pregnancies. Some probably abort in the first trimester and so go undiagnosed. However, some are discovered on US examination either routinely or because of complications such as bleeding, excessive uterine size or problems related to a high hCG level. With specialist obstetric care, 40% of such cases have continued into the third trimester and delivered live babies (Sebire et al 2002b).
Invasive moles
Invasive moles are usually diagnosed because serial urine or serum hCG measurements reveal a stable or rising hCG level in the weeks after molar evacuation. Patients may complain of persistent vaginal bleeding and lower abdominal pains and/or swelling. This may occur as a result of haemorrhage from leaking tumour-induced vasculature as the trophoblast invades through the myometrium, or because of vulval, vaginal or intra-abdominal metastases. The tumour may also involve other pelvic structures, including the bladder or rectum, producing haematuria or rectal bleeding, respectively. Enlarging pulmonary metastases or tumour emboli growing in the pulmonary arteries can contribute to life-threatening respiratory complications (Seckl et al 1991). The risk of these complications is clearly higher in patients where the initial diagnosis of a molar pregnancy was missed and who are not, therefore, on hCG follow-up.
Choriocarcinoma
Choriocarcinoma can present after any form of pregnancy, but most commonly occurs after a CM. Histological proof of choriocarcinoma is not usually obtained after a CM because of the risk of fatal haemorrhage caused by biopsy, and so it is impossible to distinguish from an invasive mole. Choriocarcinoma following a normal pregnancy or non-molar abortion usually presents within 1 year of delivery but can occur 17 years later (Tidy et al 1995). The presenting features may be similar to HM with vaginal bleeding, abdominal pain and a pelvic mass.
However, one-third of all choriocarcinomas present without pelvic symptoms but have symptoms from distant metastases. In these cases, lives can be saved by remembering to include choriocarcinoma in the differential diagnosis of metastatic malignancy (particularly in lungs, brain or liver) presenting in a woman of childbearing age. Any site may be involved, including skin (producing a purple lesion), cauda equina and the heart. Pulmonary disease may be parenchymal, pleural or may result from tumour embolism and subsequent growth in the pulmonary arteries (Savage et al 1998). Thus, respiratory symptoms and signs can include dyspnoea, haemoptysis and pulmonary artery hypertension. Cerebral metastases may produce focal neurological signs, convulsions, evidence of raised intracranial pressure, and intracerebral or subarachnoid haemorrhage. Hepatic metastases may cause local pain or referred pain in the right shoulder. Although none of these presentations are specific to choriocarcinoma, performing a simple pregnancy test or quantitative hCG assay can provide a vital clue to the diagnosis.
Infantile choriocarcinoma
Choriocarcinoma in the fetus or newborn is exceptionally rare, with approximately 30 reported cases (Blohm and Gobel 2004, Sebire et al 2005). While a primary choriocarcinoma within the infant is possible, the mother also had the tumour in 17 cases. Interestingly, the diagnosis was often made in the neonate before the mother. In all cases, the infant was anaemic and had a raised hCG level, but the site of metastasis was variable, including brain, liver, lung and skin. Only a few cases have been treated successfully with platinum chemotherapy (Johnson et al 2003
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


