Standard Nomenclature
In humans, the 22 pairs of autosomes and one pair of sex chromosomes may be affected by various abnormalities. Karyotypes are described using the International System for Human Cytogenetic Nomenclature, a standardized format agreed upon by the genetics community (Shaffer, 2009). Abnormalities fall into two broad categories—those of chromosome number, such as trisomy, and those of chromosome structure, such as a deletion or translocation. Each chromosome has a short arm, termed the “p” or petit arm, and a long arm known as the “q” arm, selected because it is the next letter of the alphabet. The two arms are separated by the centromere.
When reporting a karyotype, the total number of chromosomes is listed first, corresponding to the number of centromeres. This is followed by the sex chromosomes, XX or XY, and then by a description of any structural variation. Specific abnormalities are indicated by standard abbreviations, such as del (deletion) and inv (inversion). The affected region or bands of the p or q arms are then designated, so that the reader will know both the exact abnormality location and the way in which the chromosomal complement is abnormal. Some examples of standard karyotype nomenclature are shown in Table 13-1.
TABLE 13-1. Examples of Karyotype Designations Using the International System for Human Cytogenetic Nomenclature (2009)

 Abnormalities of Chromosome Number
Abnormalities of Chromosome Number
The most easily recognized chromosomal abnormalities are numerical. Aneuploidy is inheritance of either an extra chromosome—resulting in trisomy, or loss of a chromosome—monosomy. These differ from polyploidy, which is an abnormal number of haploid chromosome sets, such as triploidy. The estimated incidence of various numerical chromosomal abnormalities is shown in Table 13-2.
TABLE 13-2. Frequency of Numerical Chromosomal Abnormalities

Autosomal Trisomies
Trisomy accounts for approximately half of all chromosomal abnormalities. In most cases, it results from nondisjunction, which is failure of normal chromosomal pairing and separation during meiosis. Nondisjunction may occur if the chromosomes: (1) fail to pair up, (2) pair up properly but separate prematurely, or (3) fail to separate.
The risk of any autosomal trisomy increases steeply with maternal age, particularly after age 35 (Fig. 13-1). Aging is thought to break down the chiasmata that keep the paired chromosomes aligned. Oocytes are held suspended in midprophase of meiosis I from birth until ovulation, in some cases for 50 years. Following completion of meiosis at the time of ovulation, nondisjunction will result in one gamete having two copies of the affected chromosome, leading to trisomy if fertilized. The other gamete, receiving no copy of the affected chromosome, will be monosomic if fertilized. Between 10 and 20 percent of oocytes are aneuploid secondary to meiotic errors, compared with 3 to 4 percent of sperm. Although each chromosome pair is equally likely to have a segregation error, it is rare for trisomies other than 21, 18, or 13 to result in a term pregnancy. As shown in Figure 13-2, many fetuses with autosomal trisomy will be lost before term.
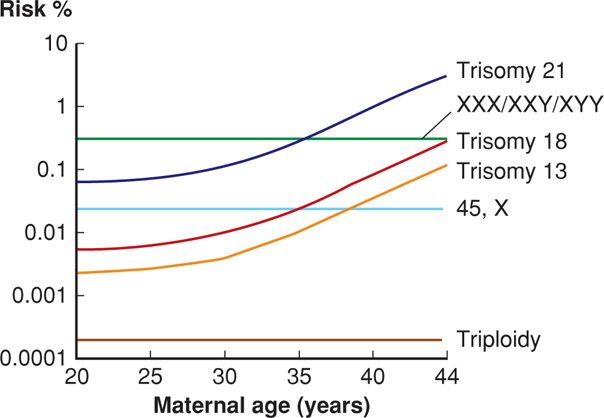
FIGURE 13-1 Maternal age-related risk for selected aneuploidies. (Redrawn from Nicolaides, 2004, with permission.)
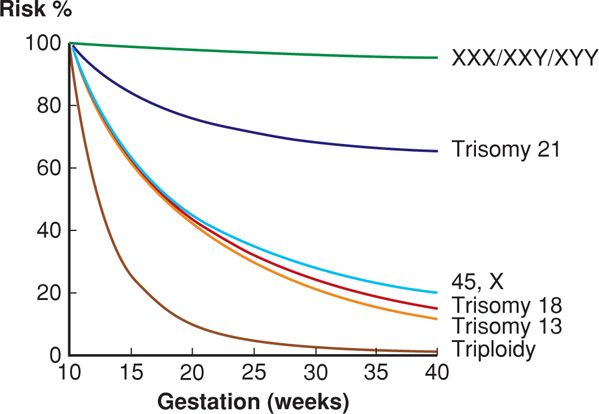
FIGURE 13-2 Gestational-age-related risk for selected chromosomal abnormalities, relative to the risk at 10 weeks’ gestation. (Redrawn from Nicolaides, 2004, with permission.)
Following a pregnancy with an autosomal trisomy, the risk for any autosomal trisomy in a future pregnancy is approximately 1 percent until the woman’s age-related risk exceeds this. Accordingly, invasive prenatal diagnosis is offered in subsequent pregnancies (Chap. 14, p. 297). Parental chromosomal studies are not indicated unless Down syndrome was due to an unbalanced translocation.
Trisomy 21—Down Syndrome. In 1866, J. L. H. Down described a group of mentally retarded children with distinctive physical features. Nearly 100 years later, Lejeune (1959) demonstrated that Down syndrome is caused by an autosomal trisomy. The trisomy 21 karyotype is shown in Figure 13-3. Trisomy 21 is the etiology of 95 percent of Down syndrome cases, whereas 3 to 4 percent is due to a robertsonian translocation. The remaining 1 to 2 percent is secondary to an isochromosome or mosaicism. The nondisjunction that results in trisomy 21 occurs during meiosis I in almost 75 percent of cases. The remaining events occur during meiosis II.
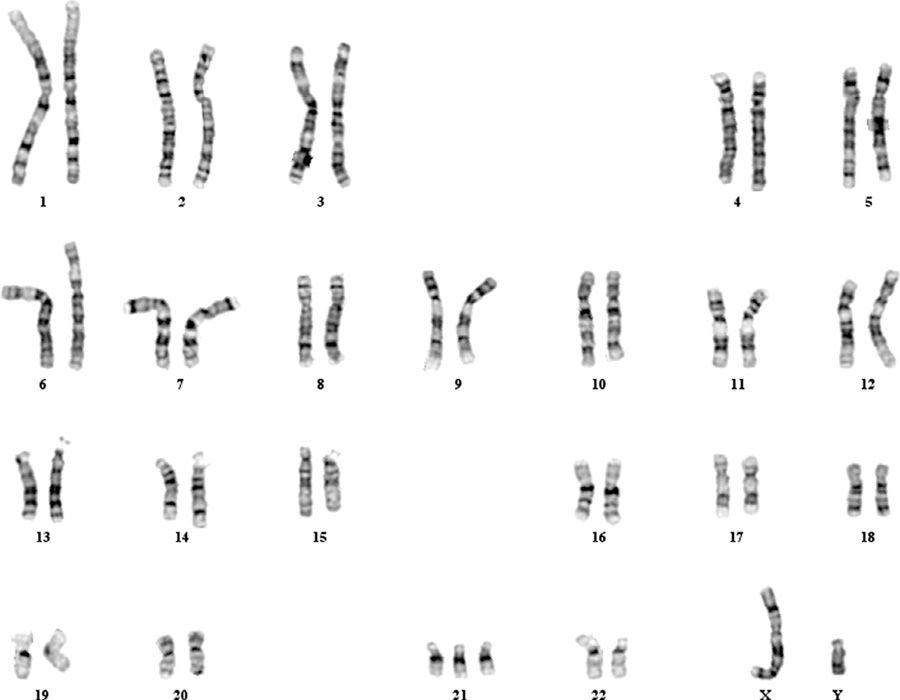
FIGURE 13-3 Abnormal male karyotype with trisomy 21, consistent with Down syndrome (47,XY,+21). (Photograph contributed by Dr. Frederick Elder.)
Down syndrome is the most common nonlethal trisomy. Its prevalence is approximately 1 per 500 recognized pregnancies, including abortuses, stillbirths, and liveborn infants (Dolk, 2010). There is a significant fetal loss rate, as shown in Figure 13-2. Approximately 30 percent of fetuses with Down syndrome are lost between 12 and 40 weeks, and 20 percent between 16 and 40 weeks (Snijders, 1999). As a result, Down syndrome is found in 1 per 740 live births in the United States or 13.5 per 10,000. This represents an increase of approximately 33 percent compared with the rate in the late 1970s (Parker, 2010; Shin, 2009). The rise in prevalence is explained by the increase in maternal age distribution during this period.
Adult women with Down syndrome are fertile, and a third of their offspring will have Down syndrome (Scharrer, 1975). Contraceptive options are discussed in Chapter 38 (p. 695). Males with Down syndrome are almost always sterile because of markedly decreased spermatogenesis.
Clinical Findings. It is estimated that 25 to 30 percent of second-trimester fetuses with Down syndrome will have a major malformation that can be identified sonographically (Vintzileos, 1995). Approximately 40 percent of liveborn infants with Down syndrome are found to have cardiac defects, particularly endocardial cushion defects and ventricular septal defects (Figs. 10-22 and 10-23, p. 210). Gastrointestinal abnormalities develop in 7 percent and include duodenal atresia, esophageal atresia, and Hirschsprung disease (Fig. 10-28, p. 214) (Rankin, 2012).
Characteristic features of Down syndrome are shown in Figure 13-4. Typical findings include brachycephaly; epicanthal folds and up-slanting palpebral fissures; Brushfield spots, which are grayish spots on the periphery of the iris; a flat nasal bridge; and hypotonia. Infants often have loose skin at the nape of the neck, short fingers, a single palmar crease, hypoplasia of the middle phalanx of the fifth finger, and a prominent space or “sandal-toe gap” between the first and second toes. Some of these findings are sonographic markers for Down syndrome, which are reviewed in Chapter 14 (p. 292).
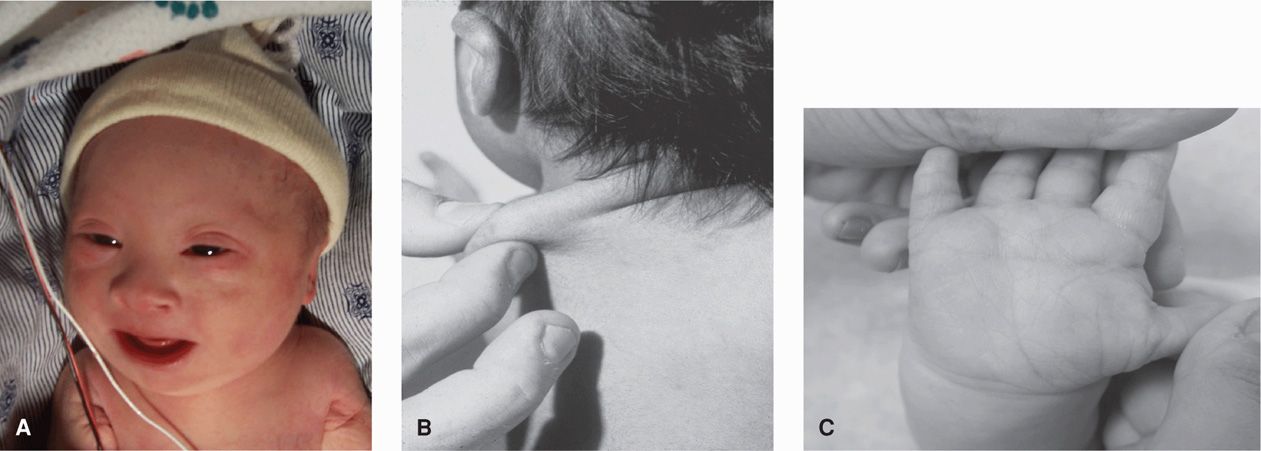
FIGURE 13-4 Trisomy 21—Down syndrome. A. Characteristic facial appearance. B. Redundant nuchal tissue. C. Single transverse palmar crease. (Photographs contributed by Dr. Charles P. Read and Dr. Lewis Waber.)
Health problems more common in children with Down syndrome include hearing loss in 75 percent, severe optical refractive errors in 50 percent, cataracts in 15 percent, thyroid disease in 15 percent, and an increased incidence of leukemia (American Academy of Pediatrics, 2001). The degree of mental impairment is usually mild to moderate, with an average intelligence quotient (IQ) score of 35 to 70. Social skills in affected children are often higher than predicted by their IQ scores.
Recent data suggest that approximately 95 percent of liveborn infants with Down syndrome survive the first year. The 10-year survival rate is at least 90 percent overall and is 99 percent if major malformations are absent (Rankin, 2012; Vendola, 2010). A number of organizations offer education and support for prospective parents faced with diagnosis of a Down syndrome fetus. These include the March of Dimes, National Down Syndrome Congress (www.ndsccenter.org), and National Down Syndrome Society (www.ndss.org).
Trisomy 18—Edwards Syndrome. This constellation of abnormalities and their association with another autosomal trisomy was first described by Edwards (1960). In population-based series, prevalence of trisomy 18 is approximately 1 per 2000 recognized pregnancies—including abortuses, stillbirths, and live births, and approximately 1 per 6600 liveborn infants (Dolk, 2010; Parker, 2010). The difference in prevalence is explained by the high in-utero lethality of the condition, as 85 percent of trisomy 18 fetuses are lost between 10 weeks’ gestation and term (see Fig. 13-2). Perhaps not surprisingly, survival of liveborn infants is likewise bleak. More than half die within the first week, and the 1-year survival rate is only approximately 2 percent (Tennant, 2010; Vendola, 2010). The syndrome is three- to fourfold more common in females (Lin, 2006; Rosa, 2011). Unlike Down and Patau syndromes, which involve acrocentric chromosomes and thus may stem from a robertsonian translocation, it is uncommon for Edwards syndrome to result from a chromosomal rearrangement.
Clinical Findings. Virtually every organ system can be affected by trisomy 18. Common major anomalies include heart defects in almost 95 percent—particularly ventricular septal defects, as well as cerebellar vermian agenesis, enlarged cisterna magna, myelomeningocele, diaphragmatic hernia, omphalocele, imperforate anus, and renal anomalies such as horseshoe kidney (Lin, 2006; Rosa, 2011; Yeo, 2003). Sonographic images of several of these are shown in Chapter 10 (p. 200).
Cranial and extremity abnormalities are also particularly common and include a prominent occiput, posteriorly rotated and malformed ears, micrognathia, small mouth, clenched hands with overlapping digits, radial aplasia, hypoplastic nails, and rockerbottom or clubbed feet. Characteristic sonographic findings include a “strawberry-shaped” cranium and choroid plexus cysts (Fig. 13-5). In otherwise low-risk pregnancies, the risk for trisomy 18 is increased only when a choroid plexus cyst is associated with other abnormalities. Alone, this cyst may be considered a normal variant.
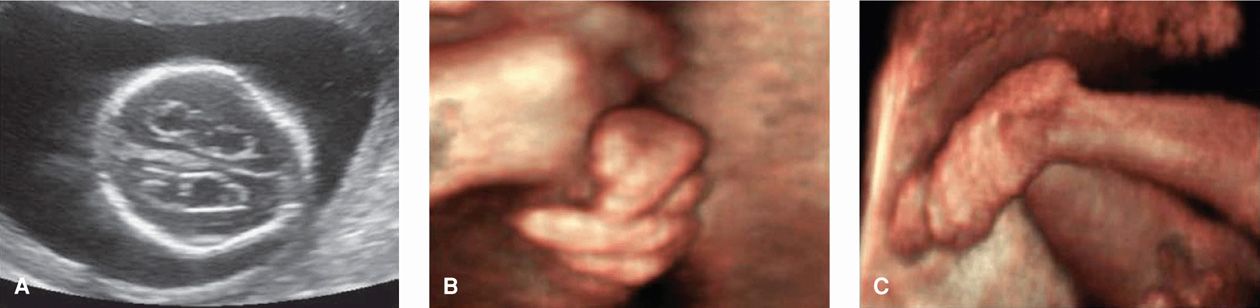
FIGURE 13-5 Trisomy 18–Edwards Syndrome. A. This transventricular sonographic view shows fetal choroid plexus cysts and a “strawberry-shaped” (unusually angulated) skull. Although not shown here, the fetal profile typically demonstrates micrognathia, with a very small, recessed mandible. B. This three-dimensional (3-D) sonographic image shows the characteristic hand position of clenched fists with overlapping digits. C. 3-D sonographic image displays a rockerbottom foot.
Pregnancies with trisomy 18 that reach the third trimester often develop fetal-growth restriction, and the mean birthweight is less than 2500 grams (Lin, 2006; Rosa, 2011). Mode of delivery should be discussed in advance, because abnormal fetal heart rate tracings are common during labor. In older reports, more than half of undiagnosed fetuses underwent cesarean delivery for “fetal distress” (Schneider, 1981).
Trisomy 13—Patau Syndrome. This constellation of fetal abnormalities and their association with yet another autosomal trisomy was described by Patau and colleagues (1960). The prevalence of trisomy 13 is approximately 1 per 12,000 live births and 1 per 5000 recognized pregnancies, which includes abortuses and stillbirths (Dolk, 2010; Parker, 2010). As with trisomy 18, trisomy 13 is highly lethal, and most affected fetuses are lost between 10 weeks and term (see Fig. 13-2).
Approximately 80 percent of pregnancies with Patau syndrome result from trisomy 13. The remainder are caused by a robertsonian translocation involving chromosomes 13 and 14, der(13;14)(q10;q10). This translocation is the most common structural chromosomal rearrangement. It is carried by approximately 1 in 1300 individuals, although the risk of an affected liveborn infant is less than 2 percent (Nussbaum, 2007).
Clinical Findings. Trisomy 13 is associated with abnormalities of virtually every organ system. One characteristic finding is holoprosencephaly. This is present in approximately two thirds of cases and may be accompanied by microcephaly, hypotelorism, and nasal abnormalities that range from a single nostril to a proboscis (Solomon, 2010). Cardiac defects are found in up to 90 percent of fetuses with trisomy 13 (Shipp, 2002). Other abnormalities that suggest trisomy 13 include neural-tube defects—particularly cephalocele, microphthalmia, cleft lip- palate, omphalocele, cystic renal dysplasia, polydactyly, rockerbottom feet, and areas of skin aplasia (Lin, 2007). For the fetus or infant with a cephalocele, cystic kidneys, and polydactyly, the differential diagnosis includes trisomy 13 and the autosomal-recessive Meckel-Gruber syndrome, which is lethal. Sonographic images of several of these abnormalities are shown in Chapter 10 (p. 201).
Few trisomy 13 fetuses survive until birth. Of those that do, the 1-week survival is approximately 40 percent, and 1-year survival is only about 3 percent (Tennant, 2010; Vendola, 2010). Counseling regarding prenatal diagnosis and management options is similar to that described with trisomy 18.
For the mother, trisomy 13 is the only aneuploidy linked with an increased risk for preeclampsia. Hyperplacentosis and preeclampsia develop in up to half of pregnancies with trisomy 13 carried beyond the second trimester (Tuohy, 1992). Chromosome 13 contains the gene for soluble fms-like tyrosine kinase-1, called sFlt-1, which is an antiangiogenic protein associated with preeclampsia. Investigators have documented overexpression of the sflt-1 protein by trisomic 13 placentas and in serum of women with preeclampsia (Bdolah, 2006; Silasi, 2011). The role of antiangiogenic growth factors in the etiopathogenesis of preeclampsia is discussed in Chapter 40 (p. 735).
Other Trisomies. In the absence of mosaicism, which is discussed below, it is rare for other autosomal trisomies to result in a live birth. There are case reports of live births with trisomy 9 and with trisomy 22 (Kannan, 2009; Tinkle, 2003). Trisomy 16 is the most common trisomy found with first-trimester losses, accounting for 16 percent, but it is not identified later in gestation. Trisomy 1 has never been reported.
Monosomy
Nondisjunction creates an equal number of nullisomic and disomic gametes. As a rule, missing chromosomal material is more devastating than having extra chromosomal material, and almost all monosomic conceptuses are lost before implantation. The one exception is monosomy for the X chromosome, Turner syndrome, which is discussed subsequently. Despite the strong association between maternal age and trisomy, there is no association between maternal age and monosomy (see Fig. 13-1).
Polyploidy
This is an abnormal number of complete haploid chromosomal sets. Polyploidy accounts for approximately 20 percent of spontaneous abortions but is rarely encountered later in gestation.
Triploid pregnancies have three haploid sets or 69 chromosomes. To have three haploid chromosomal sets, one parent must contribute two sets, and the phenotypic presentation differs according to the parent of origin. In diandric triploidy, also known as type I triploidy, the extra chromosomal set is paternal, resulting from fertilization of one egg by two sperm or by a single diploid—and thus abnormal—sperm. Diandric triploidy produces a partial molar pregnancy, which is discussed in Chapter 20 (p. 396). Diandric triploidy accounts for most triploid conceptions, but the first-trimester loss rate is extremely high. As a result, two thirds of triploid pregnancies identified beyond the first trimester are caused instead by digynic triploidy (Jauniaux 1999). In a digynic triploid pregnancy, also known as type II triploidy, the extra chromosomal set is maternal, and the egg fails to undergo the first or second meiotic division before fertilization. Digynic triploid placentas do not develop molar changes. However, the fetus often displays asymmetric growth restriction (Jauniaux, 1999).
Triploidy is a lethal aneuploidy, and more than 90 percent of fetuses with either the diandric or digynic form have multiple structural anomalies. These include abnormalities of the central nervous system, heart, face, and extremities, as well as severe growth restriction (Jauniaux, 1999). Counseling, prenatal diagnosis, and delivery options are similar to those for trisomies 18 and 13. The recurrence risk for a woman whose triploid fetus survived past the first trimester is 1 to 1.5 percent, and thus prenatal diagnosis is offered in future pregnancies (Gardner, 1996).
Tetraploid pregnancies have 4 haploid sets or 92 chromosomes. Four sets of chromosomes results in either 92,XXXX or 92,XXYY. This suggests a postzygotic failure to complete an early cleavage division. The conceptus invariably succumbs, and the recurrence risk is minimal.
Sex Chromosome Abnormalities
45,X—Turner Syndrome. This is the only monosomy compatible with life. However, it is also the most common aneuploidy in abortuses and accounts for 20 percent of first-trimester losses. The prevalence of Turner syndrome is approximately 1 per 5000 live births or 1 per 2500 girls (Cragan, 2009; Dolk, 2010). The missing X chromosome is paternally derived in 80 percent of cases (Cockwell, 1991; Hassold, 1991).
Monosomy X encompasses three distinct phenotypes. Approximately 98 percent of these conceptuses are so abnormal that they abort early in the first trimester. In a second group, large cystic hygromas are identified in either the first or second trimester, frequently accompanied by hydrops (Fig. 10-16, p. 206 and Chap. 15, p. 315). In such cases, fetal demise almost invariably results. Only the third and least-common phenotype has the potential for postnatal survival. Affected fetuses may have small cystic hygromas visible in the first or second trimester, which do not result in hydrops, and they often have other major abnormalities. One reason for the wide range of Turner syndrome phenotype is that only half of liveborn infants actually have monosomy X. Approximately one fourth have mosaicism, such as 45,X/46,XX or 45,X/46,XY. Another 15 percent have isochromosome X, that is, 46,X,i(Xq)(Milunsky, 2004; Nussbaum, 2007).
Abnormalities associated with Turner syndrome include a major cardiac malformation—such as coarctation of the aorta or bicuspid aortic valve—in 30 to 50 percent; renal anomalies, particularly horseshoe kidney; and hypothyroidism. Other features include short stature, broad chest with widely spaced nipples, congenital lymphedema, webbed posterior neck (resulting from cystic hygromas), and minor bone and cartilage abnormalities. Intelligence is generally in the normal range, although affected individuals are more likely to have visual-spatial organization deficits and difficulties with nonverbal problem solving and interpretation of social cues (Jones, 2006). Growth hormone is typically administered in childhood to ameliorate short stature (Kappelgaard, 2011). More than 90 percent have ovarian dysgenesis and require estrogen repletion beginning just before adolescence. An exception is if a mosaicism involves a Y chromosome. Such cases are at risk for germ cell neoplasm—regardless of whether the child is phenotypically male or female, and eventual prophylactic bilateral gonadectomy is indicated (Cools, 2011; Schorge, 2012).
47,XXX. Approximately 1 in 1000 female infants has an additional X chromosome—47,XXX. The extra X is maternally derived in more than 90 percent of cases (Milunsky, 2004). Pubertal development and fertility are usually normal, although premature ovarian failure has been reported (Holland, 2001). Tall stature is common. The overall major malformation rate is not increased with 47,XXX. That said, atypical phenotypic features have been described in some individuals and include epicanthal folds, clinodactyly, hypotonia, genitourinary problems, and seizure disorders (Tartaglia, 2010). Attention deficit disorder and delays in language development and motor skills have also been reported (Linden, 2002). It is estimated that because of the variability in presentation and subtlety of abnormal findings, only 10 percent of affected children are ascertained clinically.
Females with two or more extra X chromosomes—48,XXXX or 49,XXXXX—are likely to have physical abnormalities apparent at birth. These abnormal X complements are associated with varying degrees of mental retardation. For both males and females, the IQ score is lower with each additional X chromosome.
47,XXY—Klinefelter Syndrome. This is the most common sex chromosome abnormality. It occurs in approximately 1 per 600 male infants. The additional X chromosome is maternally or paternally derived with equal propensity (Jacobs, 1995; Lowe, 2001). There is also a slight association with either advanced maternal age or advanced paternal age (Milunsky, 2004).
Infants with XXY appear phenotypically normal and usually do not have an increased incidence of anomalies. As children, boys are typically tall and have normal prepubertal development. However, they have gonadal dysgenesis, do not undergo normal virilization, and require testosterone supplementation beginning in adolescence. They may develop gynecomastia. In general, IQ scores are within the normal range but slightly below those of siblings, and delays in speech, reading, and motor skills are not uncommon (Girardin, 2011).
47,XYY. This aneuploidy occurs in approximately 1 in 1000 male infants. There is no association with paternal age, anomaly rates are not increased, and there are no unusual phenotypic features. These boys tend to be tall, they have normal puberty, and fertility is unimpaired. They are at increased risk for oral and written language impairments, but intelligence is generally normal (Ross, 2009). A commonly held misconception was that XYY karyotype was associated with criminal or violent behavior. However, these early reports have been refuted.
Males with more than two Y chromosomes—48,XYYY—or with both additional X and Y chromosomes—48,XXYY or 49,XXXYY—have obvious physical abnormalities and significant mental retardation.
 Abnormalities of Chromosome Structure
Abnormalities of Chromosome Structure
Structural chromosomal abnormalities include deletions, duplications, translocations, isochromosomes, inversions, ring chromosomes, and mosaicism (see Table 13-1). Their overall birth prevalence is approximately 0.3 percent (Nussbaum, 2007). Identification of a structural chromosomal abnormality raises two primary questions. First, what phenotypic abnormalities or later developmental abnormalities are associated with this finding? Second, is evaluation of parental karyotype indicated—specifically, are the parents at increased risk to carry this abnormality? If so, what is their risk to have future affected offspring?
Deletions and Duplications
A chromosomal deletion indicates that a portion of a chromosome is missing, and a duplication means that a portion has been included twice. Deletions involving DNA segments large enough to be seen with standard cytogenetic karyotyping are identified in approximately 1 per 7000 births (Nussbaum, 2007). Common deletions may be referred to by eponyms—for example, del 5p is called cri du chat syndrome.
Most deletions and duplications occur during meiosis and result from malalignment or mismatching during the pairing of homologous chromosomes. When this happens, the misaligned segment may be deleted (Fig. 13-6). Or, if the mismatch remains when the two chromosomes recombine, it may result in a deletion in one chromosome and a duplication in the other. When a deletion or duplication is identified in a fetus or infant, the parents should be offered karyotyping to determine if either carries a balanced translocation—as this would significantly increase the recurrence risk.
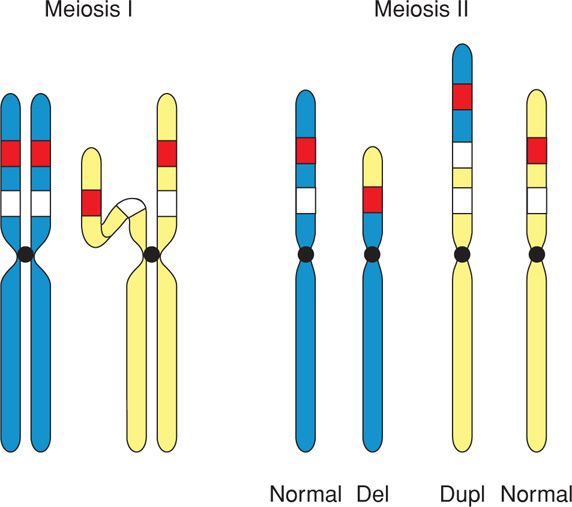
FIGURE 13-6 A mismatch during pairing of homologous chromosomes may lead to a deletion in one chromosome and a duplication in the other. del = deletion; dup = duplication.
Microdeletion Syndromes. A chromosomal deletion smaller than 3 million base pairs may not be detectable with standard karyotyping. Termed microdeletions, these may require molecular cytogenetic techniques for identification. Despite the relatively small size, a microdeletion may involve a stretch of DNA that contains multiple genes—causing a contiguous gene syndrome, which can include serious but unrelated phenotypic abnormalities (Schmickel, 1986). When a specific microdeletion syndrome is suspected, it is usually confirmed using fluorescence in situ hybridization (p. 276). Examples of common microdeletion syndromes are listed in Table 13-3.
TABLE 13-3. Some Microdeletion Syndromes Detectable by Fluorescence In Situ Hybridization (FISH)

The region of DNA that is deleted in a microdeletion syndrome (or duplicated in a microduplication) is termed a genomic copy number variant when applied to chromosomal microarray analysis discussed on page 277. Use of array-based technology has identified copy number variants that result in previously uncharacterized microdeletion syndromes—including single gene and intragenic deletions (Mikhail, 2011; Schwartz, 2012). It is likely that continued expansion of this technology will dramatically advance our knowledge of the genetic basis of disease.
22q11 Microdeletion Syndrome. This syndrome is also known as DiGeorge syndrome, Shprintzen syndrome, and velocardiofacial syndrome. It is the most common microdeletion, with prevalence of 1 per 2000 to 7000 births (Shprintzen, 2008). Although it is inherited in an autosomal dominant fashion, most cases arise from de novo mutations. The full deletion includes 3 million base pairs, encompasses 40 genes, and may include 180 different features—thus posing some counseling challenges (Shprintzen, 2008). It was once thought that different constellations of features characterized the DiGeorge and Shprintzen phenotypes, but it is now accepted that they represent the same microdeletion (McDonald-McGinn, 2011).
Associated abnormalities include conotruncal cardiac anomalies in more than 75 percent of affected individuals, such as tetralogy of Fallot, pulmonary atresia, truncus arteriosus, interrupted aortic arch, and ventricular septal defects. Immune deficiency, such as T-cell lymphopenia, also develops in approximately 75 percent. More than 70 percent have velopharyngeal insufficiency or cleft palate. Other manifestations include learning disabilities and mental retardation, hypocalcemia, renal anomalies, esophageal dysmotility, hearing loss, behavioral disorders, and psychiatric illness. Short palpebral fissures, bulbous nose tip, micrognathia, short philtrum, and small or posteriorly rotated ears are characteristic facial features (McDonald-McGinn, 2011).
Microduplication Syndromes. These syndromes are caused by duplication of DNA regions smaller than 3 million base pairs. In some cases, a microduplication may involve the exact DNA region that causes a recognized microdeletion syndrome. Examples of these include the velocardiofacial, Smith-Magenis, and Williams-Beuren syndromes (Hassed, 2004; Potocki, 2000; Somerville, 2005).
Chromosomal Translocations
These are DNA rearrangements in which a segment of DNA breaks away from one chromosome and attaches to another. The rearranged chromosomes are called derivative (der) chromosomes. There are two types—reciprocal and robertsonian translocations.
Reciprocal Translocations. A double-segment or reciprocal translocation develops when there are breaks in two different chromosomes and the broken fragments are exchanged, so that each affected chromosome contains a fragment of the other. If no chromosomal material is gained or lost in this process, the translocation is considered balanced. The prevalence of reciprocal translocations is approximately 1 per 600 births (Nussbaum, 2007). Although transposition of chromosomal segments can cause abnormalities—due to repositioning of specific genes—the balanced carrier is usually phenotypically normal. The risk of a major structural or developmental abnormality in an apparent balanced translocation carrier is approximately 6 percent. Interestingly, using microarray-based studies, as many as 20 percent of individuals who would otherwise appear to have a balanced translocation may have missing or redundant DNA segments that are below the resolution of a standard karyotype (Manning, 2010).
Balanced translocation carriers are at risk to produce unbalanced gametes that result in abnormal offspring. As shown in Figure 13-7, if an oocyte or sperm contains a translocated chromosome, fertilization results in an unbalanced translocation—monosomy for part of one affected chromosome and trisomy for part of the other. The observed risk of a specific translocation can often be estimated by a genetic counselor. In general, translocation carriers identified after the birth of an abnormal child have a 5- to 30-percent risk of having liveborn offspring with unbalanced chromosomes. Carriers identified for other reasons, for example, during an infertility evaluation, have only a 5-percent risk. This is probably because the gametes are so abnormal that conceptions are nonviable.
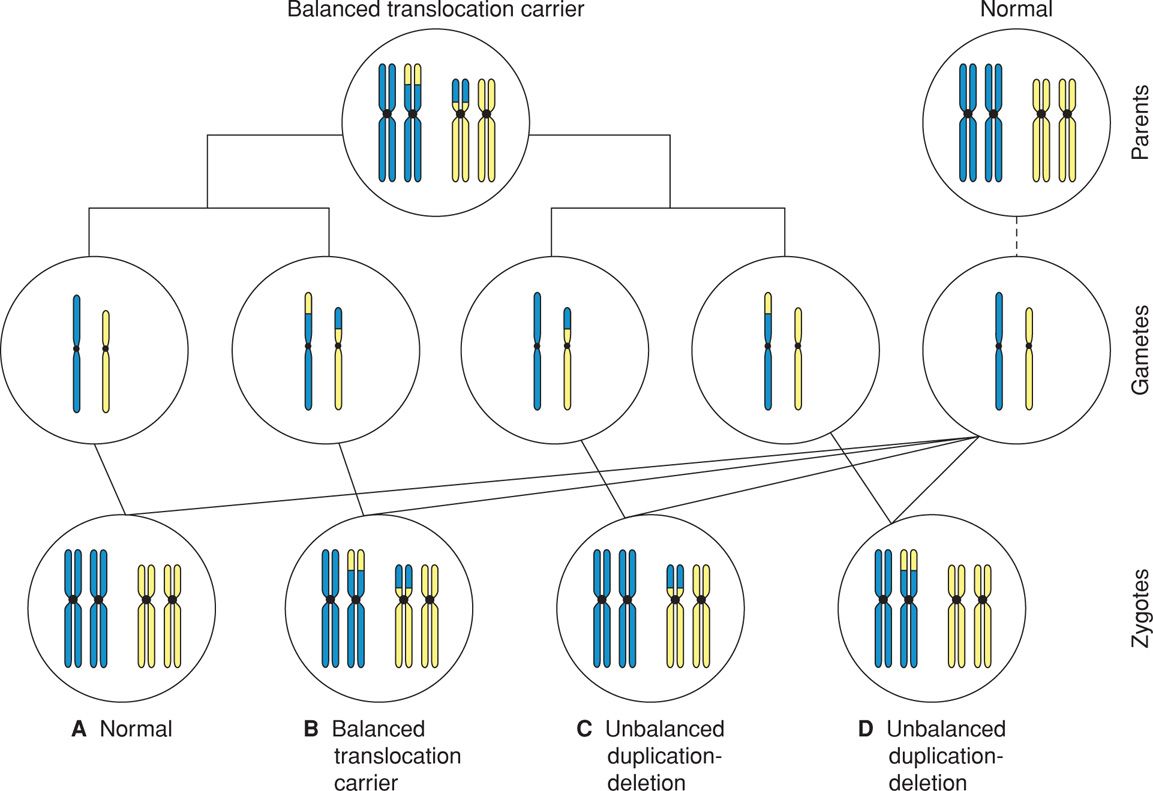
FIGURE 13-7 A carrier of a balanced translocation may produce offspring who are also carriers of the balanced rearrangement (B), offspring with unbalanced translocations (C, D), or offspring with normal chromosomal complements (A).
Robertsonian Translocations. These involve only acrocentric chromosomes, which are chromosomes 13, 14, 15, 21, and 22. In an acrocentric chromosome, the p arm is extremely short. In a robertsonian translocation, the q arms of two acrocentric chromosomes fuse at one centromere to form a derivative chromosome. Also, one centromere and the p arms of each chromosome are lost. The p arms contain the satellite regions, which contain only genes coding for ribosomal RNA. As these are present in multiple copies on other acrocentric chromosomes, the translocation carrier is usually phenotypically normal. Because the number of centromeres determines the chromosome count, a robertsonian translocation carrier has only 45 chromosomes.
Robertsonian translocations are found in approximately 1 per 1000 newborns. Balanced carriers have reproductive difficulties for a number of reasons. If the fused chromosomes are homologous, from the same chromosome pair, the carrier can produce only unbalanced gametes. Each egg or sperm contains either both copies of the translocated chromosome, which would result in trisomy if fertilized, or no copy, which would result in monosomy. If the fused chromosomes are nonhomologous, four of the six possible gametes would be abnormal.
The most common robertsonian translocation is der(13;14)(q10;q10), which may result in Patau syndrome, discussed on page 263. The observed incidence of abnormal offspring is approximately 15 percent if a robertsonian translocation is carried by the mother and 2 percent if carried by the father. Robertsonian translocations are not a major cause of miscarriage and are found in fewer than 5 percent of couples with recurrent pregnancy loss. When a fetus or child is found to have a translocation trisomy, both parents should be offered karyotype analysis. If neither parent is a carrier, the recurrence risk is extremely low.
Isochromosomes
These abnormal chromosomes are composed of either two q arms or two p arms of one chromosome fused together. Isochromosomes are thought to arise when the centromere breaks transversely instead of longitudinally during meiosis II or mitosis. They can also result from a meiotic error in a chromosome with a robertsonian translocation. An isochromosome containing the q arms of an acrocentric chromosome behaves like a homologous robertsonian translocation, and such a carrier can produce only abnormal unbalanced gametes. When an isochromosome involves nonacrocentric chromosomes, with p arms containing functional genetic material, the fusion and abnormal centromere break results in two isochromosomes. One is composed of both p arms, and one is composed of both q arms. It is likely that one of these isochromosomes would be lost during cell division, resulting in the deletion of all the genes located on the lost arm. Thus, a carrier is usually phenotypically abnormal and produces abnormal gametes. The most common isochromosome involves the long arm of the X chromosome, i(Xq), which is the etiology of 15 percent of Turner syndrome cases.
Chromosomal Inversions
When there are two breaks in the same chromosome, and the intervening genetic material is inverted before the breaks are repaired, the result is a chromosomal inversion. Although no genetic material is lost or duplicated, the rearrangement may alter gene function. There are two types—pericentric and paracentric.
Pericentric Inversion. If there are breaks in both the p and q arms of a chromosome, such that the inverted material includes the centromere, the inversion is pericentric (Fig. 13-8). This causes problems in chromosomal alignment during meiosis and confers significant risk for the carrier to produce abnormal gametes and abnormal offspring. In general, the observed risk of abnormal offspring in a pericentric inversion carrier is 5 to 10 percent if ascertainment was made after the birth of an abnormal child. However, the risk is only 1 to 3 percent if prompted by another indication. An important exception is a pericentric inversion on chromosome 9—inv(9)(p11q12), which is a normal variant present in approximately 1 percent of individuals.
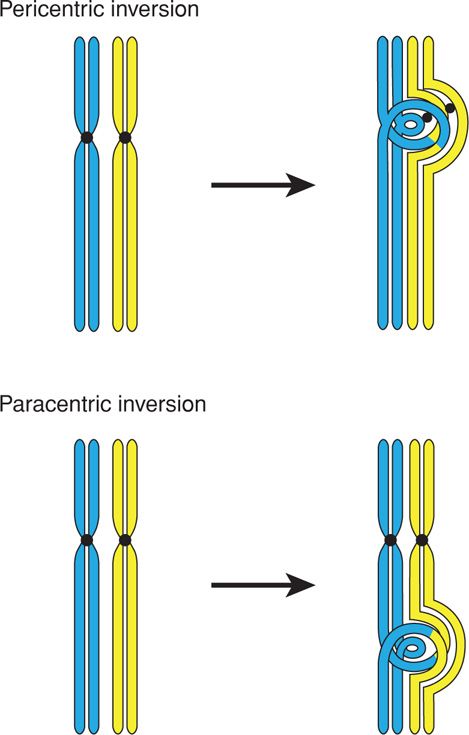
FIGURE 13-8 Mechanism of meiosis in the setting of either pericentric inversion (one involving the centromere) or paracentric inversion (not involving the centromere). Individuals with pericentric inversions are at increased risk to produce offspring with a duplication/deletion. Those with paracentric inversions are at increased risk for early pregnancy loss.
Paracentric Inversion. If there are two breaks within one arm of a chromosome, and the inverted material does not include the centromere, the inversion is paracentric (see Fig. 13-8). The carrier makes either normal balanced gametes or gametes that are so abnormal as to preclude fertilization. Thus, although infertility may be a problem, the risk of having an abnormal offspring is extremely low.
Ring Chromosomes
If deletions occur at both ends of the same chromosome, the ends may come together to form a ring chromosome. The regions at the end of each chromosome are called telomeres and contain specialized nucleoprotein complexes that stabilize chromosomes. If only telomeres are lost, all necessary genetic material is retained, so the carrier is essentially balanced. With deletions extending more proximally than the telomeres, the carrier is likely to be phenotypically abnormal. An example of this is a ring X chromosome, which may result in Turner syndrome.
Ring chromosome carriers have reproductive difficulties. The ring prevents normal chromosome alignment during meiosis and thus produces abnormal gametes. It also disrupts cell division, which may cause abnormal tissue growth and lead to short stature, borderline to moderate mental deficiency, and minor dysmorphisms. A ring chromosome may form de novo or may be inherited from a carrier parent. Parent-to-child transmission is always maternal, possibly because of compromised spermatogenesis.
Chromosomal Mosaicism
A mosaic individual has two or more cytogenetically distinct cell lines that are derived from a single zygote. Phenotypic expression of mosaicism depends on several factors, including whether the cytogenetically abnormal cells involve the placenta, the fetus, part of the fetus, or some combination. For example, mosaicism found in cells from amnionic fluid culture does not always reflect the fetal chromosome complement. The different levels of mosaicism and their clinical significance are presented in Table 13-4. When the abnormal cells are present in only a single flask of amnionic fluid, the finding is likely pseudomosaicism, caused by cell-culture artifact (Bui, 1984; Hsu, 1984). When abnormal cells involve multiple cultures, however, true mosaicism is more likely, and further testing of fetal blood or skin fibroblasts may be warranted. A second cell line is verified in 60 to 70 percent of these fetuses (Hsu, 1984; Worton, 1984).
TABLE 13-4. Types of Mosaicism Encountered in Amnionic Fluid Cultures

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


