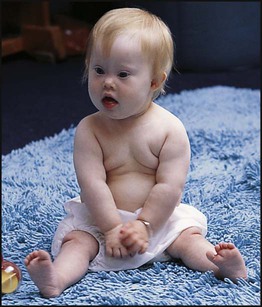
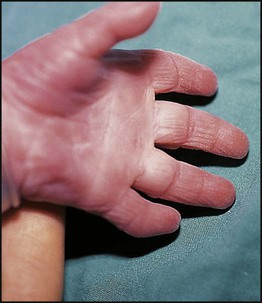
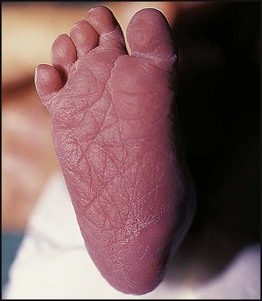
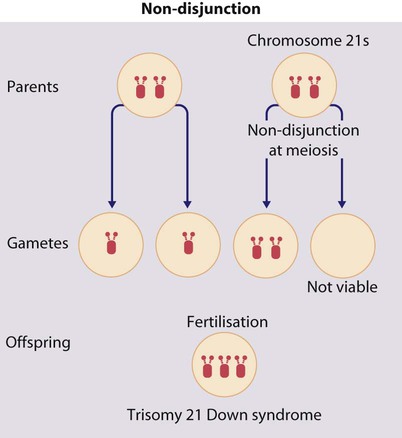
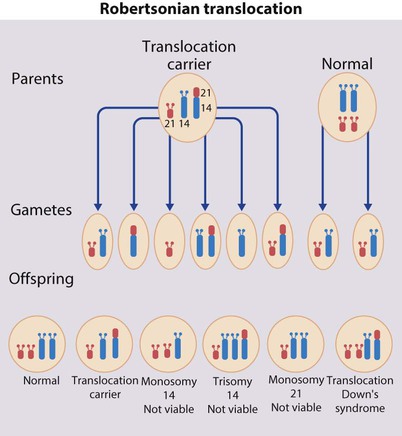
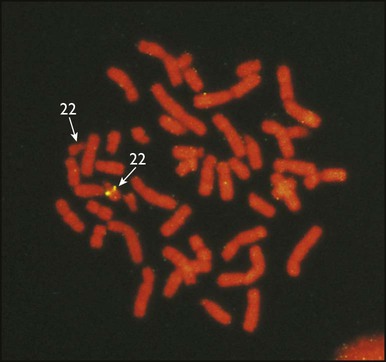
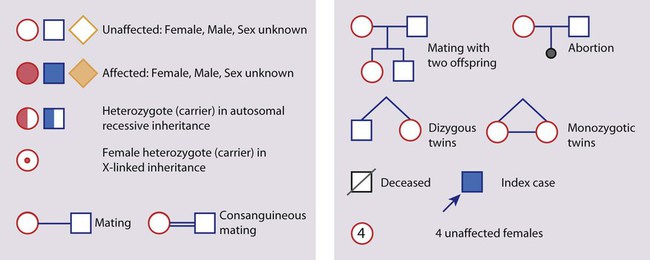
• common, with 2% of live-born babies having a significant congenital malformation and about 5% a genetic disorder • burdensome to the affected individual, family and society, as many are associated with severe and permanent disability. There has recently been an unprecedented growth in knowledge about the genetic basis of diseases : • The Human Genome Project resulted in the first publication of the human genome sequence in 2001. • It is now estimated that the human genome contains 20 000–25 000 genes, although the function of many of them remains unknown. Greater diversity and complexity at the protein level is achieved by alternative mRNA splicing and post-translational modification of gene products. • Microarray techniques and high throughput sequencing are increasing the volume and speed of genetic investigations and reducing their costs, leading to a greater understanding of the impact of genetics on health and disease. • Access to genome browser databases containing DNA sequence and protein structure has greatly enhanced progress in scientific research and the interpretation of clinical test results (Fig. 8.1). • Genetic databases are available on thousands of multiple congenital anomaly syndromes, on chromosomal variations and disease phenotypes and on all Mendelian disorders. • Clinical application of these advances is available to families through specialist genetic centres that offer investigation, diagnosis, counselling and antenatal diagnosis for an ever-widening range of disorders. • Gene-based knowledge is entering mainstream medical and paediatric practice, especially in diagnosis and in therapeutic guidance, such as for the treatment of malignancies. Genetically determined diseases include those resulting from: • the action of a single gene (Mendelian disorders) • interaction of genetic and environmental factors (polygenic, multifactorial, or complex disorders). Down syndrome is usually suspected at birth because of the baby’s facial appearance. Most affected infants are hypotonic and other useful clinical signs include a flat occiput, single palmar creases, incurved fifth finger and wide ‘sandal’ gap between the big and second toe (Fig. 8.2a–c, Box 8.1). The diagnosis can be difficult to make when relying on clinical signs alone and a suspected diagnosis should be confirmed by a senior paediatrician. Before blood is sent for analysis, parents should be informed that a test for Down syndrome is being performed. The results may take 1–2 days, using rapid FISH (fluorescent in situ hybridisation) techniques. Parents need information about the short- and long-term implications of the diagnosis. They are also likely, at some stage in the future, to appreciate the opportunity to discuss how and why the condition has arisen, the risk of recurrence and the possibility of antenatal diagnosis in future pregnancies. The extra chromosome 21 may result from meiotic non-disjunction, translocation or mosaicism. In non-disjunction trisomy 21: • most cases result from an error at meiosis • the pair of chromosome 21s fails to separate, so that one gamete has two chromosome 21s and one has none (Fig. 8.3) • fertilisation of the gamete with two chromosome 21s gives rise to a zygote with trisomy 21 The incidence of trisomy 21 due to non-disjunction is related to maternal age (Table 8.1). However, as the proportion of pregnancies in older mothers is small, most affected babies are born to younger mothers. Furthermore, meiotic non-disjunction can occur in spermatogenesis so that the extra 21 can be of paternal origin. All pregnant women are now offered screening tests measuring biochemical markers in blood samples and often also nuchal thickening on ultrasound (thickening of the soft tissues at the back of the neck) to identify an increased risk of Down syndrome in the fetus. When an increased risk is identified, amniocentesis is offered to check the fetal karyotype. After having one child with trisomy 21 due to non-disjunction, the risk of recurrence of Down syndrome is given as 1 in 200 for mothers under the age of 35 years, but remains similar to their age-related population risk for those over the age of 35 years. Table 8.1 Risk of Down syndrome (live births) with maternal age at delivery, prior to screening in pregnancy When the extra chromosome 21 is joined onto another chromosome (usually chromosome 14, but occasionally chromosome 15, 22 or 21), this is known as a Robertsonian translocation. This may be present in a phenotypically normal carrier with 45 chromosomes (two being ‘joined together’) or in someone with Down syndrome and a set of 46 chromosomes but with three copies of chromosome 21 material. In this situation, parental chromosomal analysis is recommended, since one of the parents may well carry the translocation in balanced form (in 25% of cases) (Fig. 8.4). In translocation Down syndrome: • The risk of recurrence is 10–15% if the mother is the translocation carrier and about 2.5% if the father is the carrier. • If a parent carries the rare 21 : 21 translocation, all the offspring will have Down syndrome. • If neither parent carries a translocation (75% of cases), the risk of recurrence is <1%. Although rarer than Down syndrome (1 in 8000 and 1 in 14 000 live births, respectively), particular constellations of severe multiple abnormalities suggest these diagnoses at birth; most affected babies die in infancy (Fig. 8.5, Boxes 8.2 and 8.3). The diagnosis is confirmed by chromosome analysis. Many affected fetuses are detected by ultrasound scan during the second trimester of pregnancy and diagnosis can be confirmed antenatally by amniocentesis and chromosome analysis. Recurrence risk is low, except when the trisomy is due to a balanced chromosome rearrangement in one of the parents. Usually (>95%), Turner syndrome results in early miscarriage and is increasingly detected by ultrasound antenatally when fetal oedema of the neck, hands or feet or a cystic hygroma may be identified. In live-born females, the incidence is about 1 in 2500. Figure 8.6 and Box 8.4 show the clinical features of Turner syndrome, although short stature may be the only clinical abnormality in children. • Oestrogen replacement for development of secondary sexual characteristics at the time of puberty (but infertility persists). The incidence does not increase with maternal age and risk of recurrence is very low. DiGeorge syndrome is associated with a deletion of band q11 on chromosome 22 (i.e. 22q11) (Fig. 8.7). Williams syndrome is another example of a microdeletion syndrome due to loss of chromosomal material at band q11 on the long arm of chromosome 7 (i.e. 7q11) (Fig. 8.18, see also Box 8.12). Mendelian inheritance, described by Mendel in garden peas in 1866, is the transmission of inherited traits or diseases caused by variation in a single gene in a characteristic pattern. These Mendelian traits or disorders are individually rare but collectively numerous and important: over 6000 have been described. For many disorders, the Mendelian pattern of inheritance is known. If the diagnosis of a condition is uncertain, its pattern of inheritance may be evident on drawing a family tree (pedigree), which is an essential part of genetic evaluation (Fig. 8.8). This is the most common mode of Mendelian inheritance (Box 8.6). Autosomal dominant conditions are caused by alterations in only one copy of a gene pair, i.e. the condition occurs in the heterozygous state despite the presence of an intact copy of the relevant gene. Autosomal dominant genes are located on the autosomes (chromosomes 1–22) so males and females are equally affected. Each child from an affected parent has a 1 in 2 (50%) chance of inheriting the abnormal gene (Fig. 8.9a, b). This appears to be straightforward, but complicating factors include the following factors.
Genetics
Chromosomal abnormalities
Down syndrome (trisomy 21)
Clinical features
Cytogenetics
Meiotic non-disjunction (94%)
Maternal age (years)
Risk of Down syndrome
All ages
1 in 650
20
1 in 1530
30
1 in 900
35
1 in 385
37
1 in 240
40
1 in 110
44
1 in 37
Translocation (5%)
Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13)
Turner syndrome (45, X)
Deletions
Mendelian inheritance
Autosomal dominant inheritance