120 Genetic Syndromes
One of the specific approaches to diagnosis of genetics syndromes is the incorporation of major malformations (e.g., cleft lip or palate, brain or cardiac anomalies) as well as more common minor malformations (e.g., downslanting palpebral fissures, flat nasal bridge, syndactyly, or clinodactyly; see Chapter 116) to lead the diagnostician toward a specific diagnosis. The goal of this chapter is to illustrate a systematic approach using categories of major or unique findings. Only one or two of the more common or classic syndromes are illustrated for each major category.
Very Small or SHORT Stature
Cornelia de Lange Syndrome
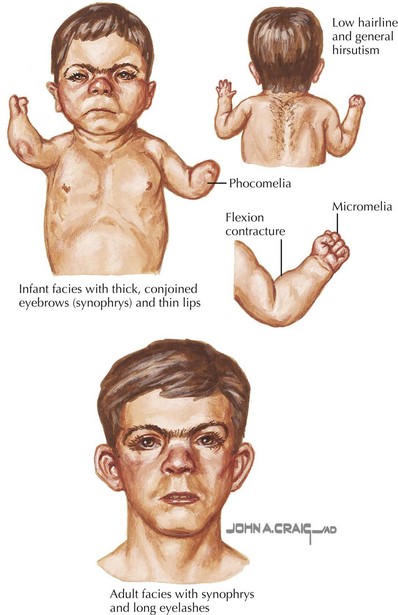
Cornelia de Lange syndrome (CdLS) has a prevalence of around 1 : 10,000 and is characterized by prenatal and postnatal growth retardation, hirsutism, and upper limb reduction defects. Craniofacial abnormalities include synophrys, arched eyebrows, long eyelashes, a small, upturned nose, small, widely spaced teeth, and microcephaly (Figure 120-1). Patients have moderate to severe mental retardation (MR), and many show autistic and self-destructive behaviors. Upper limb abnormalities range from severe deficiencies to merely proximally placed thumbs or fifth digit clinodactyly. Patients classically have growth failure as well as feeding difficulties, which include regurgitation, projectile vomiting, and swallowing difficulties. Hearing loss and speech delay are common. CdLS has dominant inheritance usually caused by de novo mutations in NIPBL (50%), SMC1A (5%), and SMC3 (<1%).
Russell-Silver Syndrome
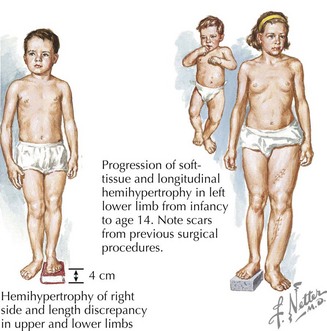
Russell-Silver syndrome (RSS) has a prevalence of about 1 : 100,000 and includes intrauterine growth retardation, postnatal growth deficiency, delayed bone age, normal head circumference, limb-length asymmetry, and fifth digit clinodactyly (Figure 120-2). A typical face in RSS is small and triangular with a broad and prominent forehead, downturned corners of the mouth, and micrognathia. Café-au-lait spots are sometimes present, and patients can sweat extensively. Nearly 40% of cases have been shown to have abnormal imprinting of chromosome 11p15.5, and 10% of cases demonstrate uniparental disomy (UPD) for chromosome 7. Growth hormone may benefit patients for whom growth plateaus early.
Premature Aging
Hutchinson-Gilford Progeria Syndrome
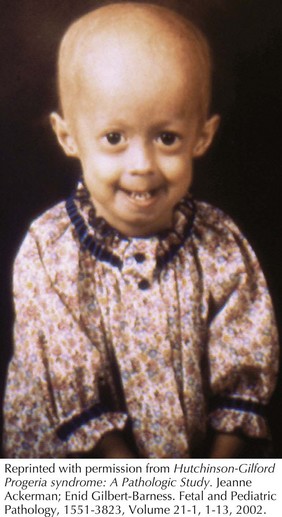
Hutchinson-Gilford Progeria syndrome (HGPS) occurs in between 4 and 8 million births and is a composite of features that develop in childhood that are reminiscent of accelerated aging (Figure 120-3). These include alopecia, diminished subcutaneous fat, stiffness of joints, bone changes, and an abnormal tightness of the skin over the abdomen and upper thighs that emerge during the second to third year of life. Facial features include a pinched nose, thin lips, protruding ears, and a thin, high-pitched voice. Skeletal manifestations include distal phalangeal osteolysis, delayed fontanelle closure, a pear-shaped thorax, micrognathia, short dystrophic clavicles, a “horse-riding” stance, thin limbs, and tightened joint ligaments. Cognitive development is normal. Patients typically develop severe atherosclerosis requiring electrocardiography, echocardiography, and carotid duplex scans every 6 months. Yearly lipid profiles and medical treatment for cardiovascular disease and elevated lipids are recommended. Hip dislocations and contractures are common and should be managed conservatively with physical and occupational therapy. Classical HGPS is caused by de novo dominant splice site mutations in the LMNA gene for which genetic testing is available.
Overgrowth Syndromes
Fragile X Syndrome
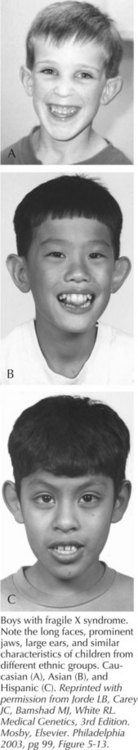
Fragile X syndrome (FXS) is seen in patients with a FMR1 full mutation and is characterized by moderate MR in males and mild MR in females. Males are macrocephalic, with long faces, prominent foreheads and chins, protruding ears, and large testes after puberty (Figure 120-4). Developmental milestones are delayed, and behavioral features such as tactile defensiveness, poor eye contact, perseverative speech, difficulty with impulse control, and distractibility become more prominent with age. More than 99% of patients with FXS have a loss-of-function mutation in the FMR1 gene at Xq27.3 that results from more than 200 repeats of a CGG tri-nucleotide (normal 0-40 repeats) and subsequent aberrant methylation of the gene. Individuals with 59 to 200 trinucleotide repeats are considered to have an FMR1 premutation and can have late-onset, progressive cerebellar ataxia and intention tremor in males and premature ovarian failure in females but with normal appearance and intellect. FXS demonstrates a phenomenon known as anticipation, in which the trinucleotide repeat becomes larger and the phenotype more pronounced with passage from one generation to the next. Molecular epidemiology has suggested 16 to 25 in 100,000 males affected and about half of that prevalence for females.
Beckwith-Wiedemann Syndrome
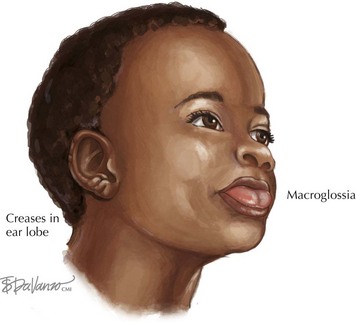
Beckwith-Wiedemann Syndrome (BWS) is a growth disorder characterized by macrosomia, macroglossia, visceromegaly, omphalocele, neonatal hypoglycemia, ear creases or pits, and a risk for embryonic tumors that includes hepatoblastoma and Wilms’ tumors (Figure 120-5). Partial overgrowth, or hemihyperplasia, may occur in tissues, organs, or body segments (see Figure 120-2). Nearly 60% of people with BWS have methylation abnormalities or mutations in the imprinted 11p15 region. Mutations in the CDKN1C gene, a growth-regulating gene in this region, are seen in 40% of familial cases and 5% to 10% of nonfamilial cases of BWS. Methylation abnormalities in KCNQ1OT1 are present in 50% to 60% of the cases and in H19 in 2% to 7% of the cases. Because of the risk of tumors, abdominal ultrasonography is recommended every 3 months until age 8 years, and serum α-fetoprotein concentration is monitored during the first 3 years.
Neurologic Findings
Prader-Willi Syndrome and Angelman Syndrome
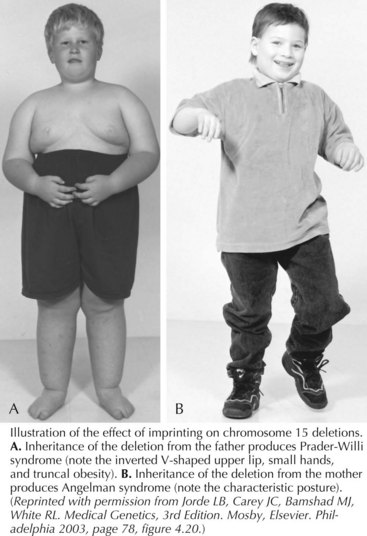
Prader-Willi syndrome (PWS) is characterized by hypotonia and difficulty feeding early in infancy followed by hyperphagia, beginning from 12 months to 6 years, that leads to central obesity. The face has a narrow bifrontal diameter, almond-shaped eyes, and a downturned mouth (Figure 120-6). Motor and language development is delayed because of cognitive impairment. Hypogonadism with incomplete pubertal development and infertility occurs. Behavioral outbursts (temper tantrums, obsessive-compulsiveness, stubbornness, and manipulation), sleep disturbances, short stature, hypopigmentation, small hands and feet, skin picking, and articulation difficulties are also seen. Treatment with growth hormone is recommended for those with PWS, and patients may thrive in settings where behavior and food intake are closely monitored.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


