40 Genetic Disorders
Cellular and Molecular Genetics
Genes, which carry the information about inherited characteristics from parent to child, are arranged linearly on the chromosomes, each with a specific locus. Thousands of genes are located on each chromosome. Not all genes are active at once; certain mechanisms activate them at various developmental points. In homozygous loci, the genes from a pair of chromosomes carry similar instructions regarding the trait of interest; in heterozygous loci, the instructions are different for each gene. In the latter case, one gene may be dominant, with its instructions manifested in the phenotype, as in Huntington disease, or the genes can be co-dominant, as in the case of individuals with blood type AB.
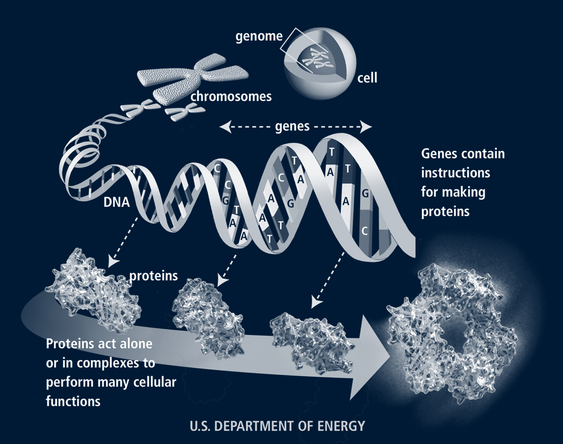
The human genome contains approximately 25,000 to 30,000 genes. Genes are composed of DNA. Each DNA molecule includes pairs of nitrogenous bases—adenine, cytosine, guanine, and thymine (labeled A, C, G, T)—wound around a histone protein core in a double helix. There are more than 3 billion base pairs in the human genome for an individual. From the four nitrogenous bases, 64 triple-base combination sequences (codons) of A, C, G, and U (uracil is substituted for thymine in the messenger ribonucleic acid [mRNA] at this point) such as GUA, UUG, and CGG are possible (Jorde et al, 2006). Three codons signal the end of a gene (stop codons) and 61 define the 20 amino acids. Thus each amino acid may be specified by more than one codon. The sequence of the amino acids directs the synthesis of proteins in the cell cytoplasm (Fig. 40-1). Telomeres at the tips of each chromosome protect the chromosome from breaking down. These deteriorate with age.
 Epigenetics
Epigenetics
There are several recognized mechanisms for epigenetic inheritance, not all of which are discussed here. DNA methylation is the most studied. When DNA material is methylated, the gene is turned off. Methylation is involved with cellular differentiation in utero but the same process occurs throughout life. A second mechanism for epigenetic modifications to DNA functioning involves the way DNA attaches to histones. It is called chromatin modification. DNA is wrapped around a series of proteins called histones. If these proteins are tightly joined to the DNA, the DNA is hidden from exposure and cannot express itself. If the histone wrap is loosened, often through acetylation, enzymes, or certain forms of RNA, then the gene may be expressed (Fig. 40-2). In general, when chromatin is tightly folded, gene expression is restricted while more open chromatin allows gene expression. In other words, gene regulation varies depending on the type of histone linkage. Certain RNA proteins are also transmitted with reproduction and are involved with epigenetic inheritance (Jablonka and Raz, 2009).
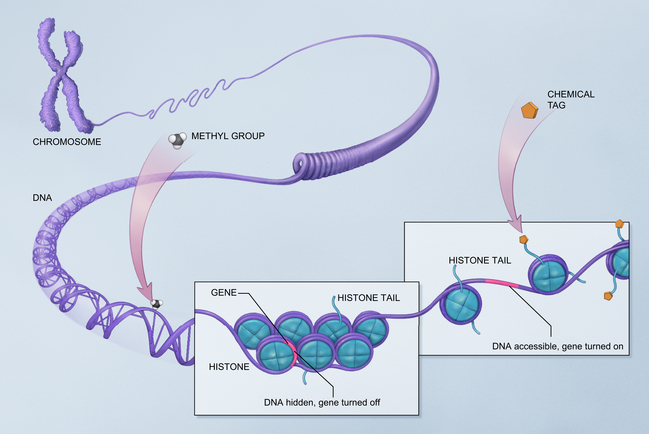
The study of epigenetics is revolutionizing our understanding of many conditions (e.g., autism spectrum disorders, which are known to have both genetic and environmental etiologic factors; epigenetics may explain how the various factors interrelate to cause these disorders). Rett, fragile X, Prader-Willi, Angelman, and all demonstrate dysregulation of normal epigenetic mechanisms (Grafodatskaya et al, 2010). It is thought that many cancers may result from epigenetic control of gene expression rather than defective genes themselves, or may be caused by a mixture of the two factors. Cancer epigenetics may involve disruption of the stem cells, sometimes when they have replicated many times over years and no longer seem to be able to function properly (Feinberg, 2007; NOVA, 2007). Some of the endocrine disruptor toxins in the environment cause epigenetic trangenerational effects, meaning that the germline is affected and this change is transmitted to future generations (Guerrero-Bosagna and Skinner, 2009).
 Ethical Issues
Ethical Issues
• Identification of the genetic condition must provide a clear benefit to the child.
• A system must be in place to confirm the diagnosis.
• Treatment and follow-up must be available for affected infants.
 Genetics Testing in the Future
Genetics Testing in the Future
Genetics testing will become more complex in the future and primary care providers will need to be knowledgeable in all these areas. First, universal screening can be expected to expand as therapies evolve, making it possible to prevent or modify significant pathology in affected individuals. Second, screening will be expanded for diagnosis of common conditions. For example, more than 50% of sensorineural hearing loss is due to genetic conditions. When infants are identified through newborn hearing screening, they will then undergo genetic testing to ascertain possible causes. Some autism and obesity cases are now connected to certain genetic profiles in children. Hypertension, diabetes, heart disease, and cancer are adult conditions in which greater genetic information will affect identification of and management of those at risk (Lose, 2008). Third, additional screening will be done to identify those at risk for future problems, such as the breast cancer and Parkinson disease tests that are already available. Finally, pharmacogenetics will involve genetic testing to ascertain drug responses in individuals with different genetic profiles (Cheng et al, 2008). Primary care providers will serve as translators for patients needing and receiving genetic information over their lifetime (Lose, 2008).
 Chromosomal Disorders
Chromosomal Disorders Single-Gene Disorders
Single-Gene Disorders Multifactorial or Multiple-Gene Disorders
Multifactorial or Multiple-Gene Disorders Nontraditional Inheritance
Nontraditional Inheritance
Three additional patterns of transmission of genetic material from generation to generation have been identified—germline mosaicism, uniparental disomy, and mitochondrial inheritance.
 Teratogens
Teratogens
Although not strictly genetic in origin, teratogens are often discussed with genetic disorders because the differential diagnosis includes factors that affect the embryo after fertilization and those that affect the DNA of the germ cells or their joining with fertilization. Fetal alcohol syndrome is an example of a condition in this category. Viral diseases, such as rubella, certain drugs, and environmental toxins—such as mercury—are also considered teratogens. The Pregnancy Exposure InfoLine (www.thepeil.org) contains current information on potential teratogenic effects of specific environmental substances.
Assessment
Primary care providers identify possible genetic disorders by using the same skills as those for other pediatric health problems: knowledge of risk factors, collection of a good history, and a complete physical examination augmented with appropriate laboratory or other studies. After the assessment, providers determine the operative genetic mechanism and develop and implement a plan of care for the patient and family with consideration of individual, family, and cultural factors. Box 40-1 identifies some common features of children or family members with genetic disorders that should lead providers to explore issues of possible genetic problems.
 Risk Factors
Risk Factors
Risk factors that may be identified in the child or family members include the following:
• Family history of known genetic disorder or recurrent pathologic condition
• Delayed development of secondary sex characteristics
• Affective disorders (e.g., schizophrenia)
• Developmental delays or learning problems
• Repeated spontaneous abortions or stillbirths
• Maternal factors, including alcohol or drug exposure, medication exposure, age older than 35 years, environmental or occupational toxin exposure
TABLE 40-1 Genetic Risks Associated With Ethnic Background
| Ethnic Background | Genetic Disorder at Higher Risk |
|---|---|
| Northern European | Cystic fibrosis, phenylketonuria |
| Jewish (Ashkenazi descent) | Tay-Sachs, Canavan, Gaucher |
| West African | Sickle cell, sickle cell–hemoglobin C |
| Mediterranean | Beta-thalassemia, sickle cell |
| French-Canadian | Tay-Sachs, branched-chain ketoaciduria |
 History
History
The history of genetic diseases usually includes the following main areas (Dolan and Moore, 2007):
• Family history of the disease using a pedigree format. A family history is needed to identify family members with conditions that may be genetically transmitted. The pedigree provides a visual map of the occurrence of specific traits and identifies other family members who might be at risk. Providers can see the potential pattern of inheritance and the relationships among affected family members. Past and current health of each person in the pedigree, birth histories of other family members, and mental retardation or learning problems of family members are all important areas to explore. Consanguinity should be noted; persons who have a common ancestor may each be carriers of a gene mutation present in that family.
• Environmental and occupational history. The environmental and occupational history may provide information about specific teratogenic factors that might be involved.
• Reproductive history. The mother’s reproductive history may give information about malformations, genetic conditions, or infectious diseases transmitted to other offspring. Her pregnancy and delivery of the child in question may give other information to determine whether the condition was the result of genetic factors or whether the condition was a result of trauma, infection, or some other factor occurring during the pregnancy or delivery.
• Medical history of the child
Figures 40-3 and 40-4 illustrate the pedigree notation format. Screening questions for genetic disorders that should be asked of all patients are included in Table 40-2. Families can be encouraged to record their own family history information by using programs such as the U.S. Surgeon General’s Family History Initiative (USDHHS, 2005). Parents and children should be encouraged to learn about the health of members of their family, which may, in turn, provide clues as to specific health risks for themselves. When a genetic disorder is suspected, the history must become more specific, as outlined in Table 40-3.
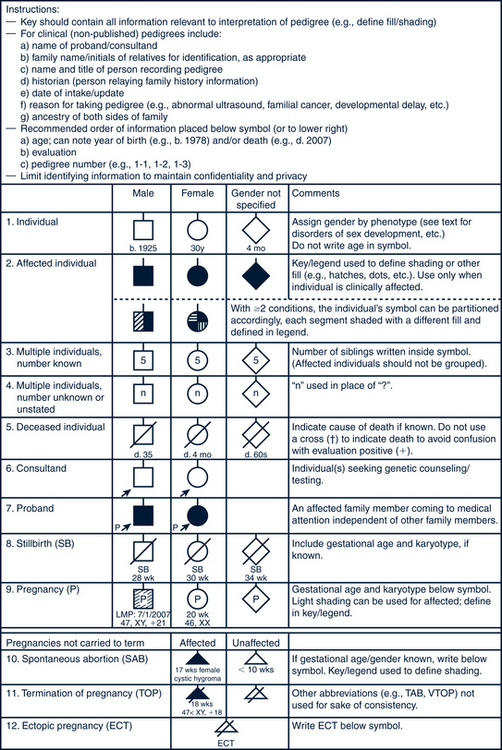
FIGURE 40-3 Pedigree model. Common pedigree symbols, definitions, and abbreviations.
(Adapted from Bennett R, French KS, Resta RG: Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors, J Genet Counsel 17:424-433, 2008.)
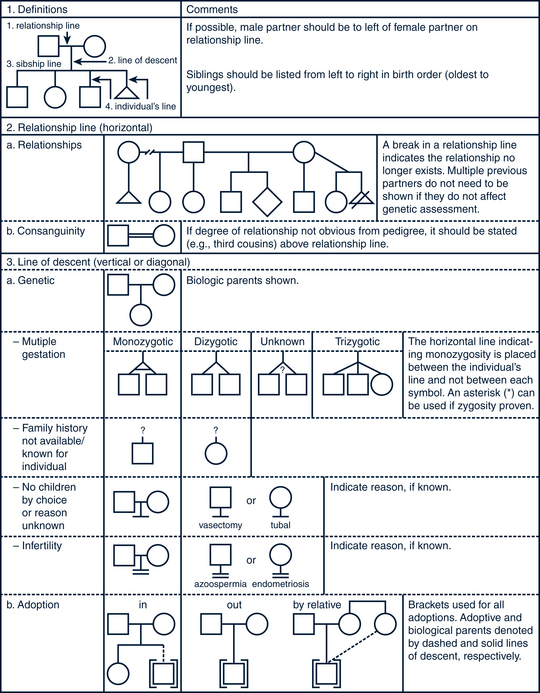
FIGURE 40-4 Pedigree line definitions.
(Adapted from Bennett R, French KS, Resta RG: Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors, J Genet Counsel 17:424-433, 2008.)
TABLE 40-2 General Screening for Genetic Conditions: The History
| Question | Rationale/Comments |
|---|---|
| Has anyone in the family had a birth defect? | To identify conditions that affect others in the family. If answer is yes, try to get more information about the nature of the defect. |
| Is there anyone in the family with a stillborn baby or baby who died early in life? | To identify unrecognized genetic disorders. Babies who died very early may have inheritable metabolic disorders. Distinguish from sudden infant death syndrome. |
| Is there any chance that you and your partner are blood-related? Is this pregnancy a product of incest? | Consanguinity of partners closer than first cousins is a risk factor for autosomal recessive disorders. If yes, recommend genetics consultation. |
| Are there any diseases or traits that run in your family? | Significant if early onset, two or more close relatives affected. Genetic heart disease and genetic cancer risks are important. If yes, recommend genetic consultation and monitoring. |
| Have you or any of your parents or siblings had three or more miscarriages? | May indicate a chromosome translocation. If yes, order a karyotype of the mother or father (or both). |
| Does anyone in the family have mental retardation? | Look for multiple members affected and associated with dysmorphic features. If yes, recommend genetic consultation. |
| What is your ethnic background? Your partner’s? | Consider ethnic risk factors and screen if at risk. |
TABLE 40-3 Specific Genetic History Questions



