Fig. 18.1
G Protein-coupled receptor activation and inactivation pathway. a In its inactive state, the Gsα is bound to guanosine diphosphate (GDP) and βγ subunits. b Agonist binding to the receptor results in conversion of GDP to hydrolyze guanosine triphosphate (GTP). c Gsα bound to GTP is in its active form and activates adenylyl cyclase, which generates cAMP. d GTPase inactivates Gsα by converting GTP to GDP. Gsα then reassociates with βγ subunits. * In FD and macrophage activation syndrome (MAS), GTPase is mutated, leading to constitutive activation of Gsα
In bone, constitutive receptor activation results in increased expression of c-fos proto-oncogene and increased interleukin-6 production, leading to abnormal proliferation and differentiation of osteoblast precursors [6, 7]. These poorly differentiated osteoblasts generate abnormal fibrous tissue in the bone marrow space. There is also activation of nearby osteoclasts, increasing bone resorption that further alters bone architecture at the site of the lesion [7]. The net result of these alterations of bone formation and resorption is the proliferation and accumulation of fibrous tissue replacing normal bone in cyst-like spaces. Lesions can occur in a single discrete site (monostotic FD), or involve multiple bones (polyostotic FD). In some patients, FD lesions also produce fibroblast growth factor 23 (FGF-23), leading to excessive loss of urinary phosphate that can complicate this disorder [8]. Renal phosphate wasting can result in concomitant osteomalacia, further weakening bones and contributing to fracture risk [9].
This constitutive receptor activation can also affect tissues other than bone. Within skin, the constitutive activation of melanocytes leads to hyperpigmented macules, termed café au lait macules (Fig. 18.2). Unlike the smooth borders of café au lait macules seen in neurofibromatosis, the characteristic macules are distinguished by their more irregular borders, sometimes referred to as “Coast of Maine” lesions, with the borders resembling the ragged Maine coastline.
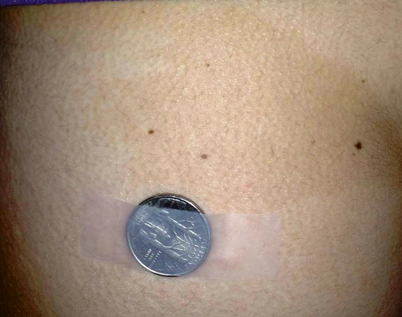
Fig. 18.2
Café au lait macule. Classic “coast of Maine” appearance marked by irregular borders
Endocrine organs may also be involved in cases of FD, with the combination of FD with café au lait macules and endocrinopathies referred to as the McCune–Albright syndrome [10]. The most well known endocrinopathy associated with McCune–Albright syndrome is gonadotropin-independent, precocious puberty caused by primary ovarian or testicular hyperfunction. Pituitary gland involvement can lead to acromegaly, Cushing’s disease, and nonfunctioning pituitary adenomas. Constitutive activation of the thyrotropin (TSH) receptor can lead to hyperthyroidism and/or thyroid nodules. Primary adrenal involvement manifests as adrenal nodules and/or hypercortisolemia (Table 18.1).
Table 18.1
Endocrine tissues expressing GNAS receptors and the resulting clinical manifestations of constitutive activation
Bone | Adrenal glands |
Fibrous dysplasia | Adrenal nodules |
FGF-23 overproduction leading to renal phosphate wasting | Cushing’s syndrome |
Ovarian/testicular tissue | Pituitary gland |
Precocious puberty | Acromegaly |
Testicular mass (Leydig and/or Sertoli cell hyperplasia) | Hyperprolactinemia |
Cushing’s disease | |
Pituitary adenoma | |
Thyroid | Parathyroid glands a |
Thyroid nodules | Primary hyperparathyroidism |
Hyperthyroidism |
FD can rarely occur in combination with benign intramuscular myxomas without associated endocrinopathies, referred to as Mazabraud’s syndrome.
Epidemiology
The prevalence is difficult to estimate, but FD has been reported to comprise up to 7 % of all benign bone tumors [11]. Because this mutation occurs post-zygotically, FD and the McCune–Albright syndrome are noninheritable. Males and females tend to be similarly affected . Approximately 60 % of affected patients have the monostotic form of FD, which typically presents during young adulthood . The less common polyostotic form tends to be more severe and presents at a younger age, often in children under 10 years of age . Craniofacial bones are affected in approximately 10 % of cases of monostotic FD and more than 50 % of cases of polyostotic FD. McCune–Albright syndrome occurs in a small subset of patients with polyostotic FD, comprising less than 5 % of all affected patients [12–15].
Clinical Presentation
The clinical presentation of FD includes a spectrum, ranging from asymptomatic to severe depending on the number and anatomical location of affected sites. Any bone can be involved, although the femur and skull base are the most commonly affected. FD may be incidentally discovered as a bony lesion found on unrelated radiology images. Patients may also present with bone pain or tenderness corresponding to the affected region. If the long bones are affected, patients may also develop an abnormal skeletal angulation that is often referred to as a “shepherd’s crook” deformity. In addition, patients can present with a pathologic fracture at the site of the FD lesion due to weakening of the bone from an abnormal architecture. In one study, peak fracture rate occurred between the ages of 6 and 10 years, with a decline in rate of fracture thereafter [9]. Craniofacial involvement can lead to asymmetry and disfigurement of affected areas, as well as abnormal tooth development and jaw issues. Compression of important intracranial structures can cause visual impairment or cranial nerve palsies. Scoliosis can be a presenting sign of spine involvement [16]. The McCune–Albright syndrome can present either with the above bone manifestations or from effects of associated endocrinopathies, most commonly precocious puberty.
Differential Diagnosis
FD lesions display characteristic findings on imaging as described in more detail below, and few other disease processes produce a similar clinical picture. Paget’s disease of bone, a disease characterized by expansive bony lesions, can occur in one or more discrete bony regions and result in expansion of the cortical envelope similar to FD. These lesions are distinct from FD lesions in that they typically occur later in life and can typically be distinguished on imaging. Solitary lesions seen in monostotic FD can resemble other bony tumors or processes . For example, simple bone cysts, nonossifying fibromas, osteofibrous dysplasia, bone angiomas, and low-grade, intramedullary osteosarcoma or other sarcomas can resemble an isolated FD lesion. In addition, craniofacial FD lesions may appear similar to a calcified meningioma. In cases where imaging does not confirm the diagnosis, biopsy of the lesion can be performed [17, 18].
Diagnosis and Evaluation
Physical Examination
Distinct findings on physical examination may be subtle or absent. Bone deformities, such as abnormal long bone angulation or asymmetric craniofacial swelling, may be identified on close inspection. Tenderness to palpation of affected bones may be present. In craniofacial FD, impingement of cranial nerves can result in visual abnormalities or cranial nerve palsies. Consultation with an ophthalmologist for regular complete ophthalmologic examinations is indicated in craniofacial cases. Careful skin examination may reveal café au lait macules. In the McCune–Albright syndrome, evidence of hormonal excess may also be apparent on exam . Palpable masses within the thyroid or testis may be noted, or the clinical stigmata of Cushing’s syndrome, acromegaly, or hyperthyroidism. Lastly, premature pubertal development in children may be noted if precocious puberty is present.
Laboratory Evaluation
In monostotic and polyostotic FD, laboratory evaluation may reveal abnormalities in bone turnover, with increased markers of bone resorption and formation. Serum alkaline phosphatase may also be elevated due to an increase in bone-specific alkaline phosphatase . Electrolytes, calcium, and renal function are typically normal. If excess FDF-23 is produced by FD lesions, phosphate levels may be low, associated with high urinary phosphate levels and low tubular resorption of phosphate (TRP) due to renal phosphate wasting. Serum 25-hydroxyvitamin D levels should be measured, since previous studies have shown that vitamin D deficiency is not uncommon in this patient population [19].
All patients with polyostotic FD should also undergo evaluation for underlying endocrinopathies to evaluate for McCune–Albright syndrome, including evaluation for hyperthyroidism, cortisol excess, and acromegaly. In prepubertal children, this evaluation should also include an assessment for physical or biochemical evidence of precocious puberty as suggested by the history, physical examination or results of laboratory tests [20].
Imaging
Findings on imaging studies can be diagnostic of FD. Appearance on plain radiographs can vary, but typically reveal expansion of bone from the medullary space outward to the cortex in discrete well-defined regions. Cortices are often thinned and can appear sclerotic. The center of the lesion may have a radiolucent or ground glass appearance. The lesions can develop an increasingly sclerotic appearance with advancing age. Associated deformities can be seen in long bones, including abnormal angulation or bowing [21]. In polyostotic forms, a hemimelic distribution of lesions may often, although not always, be seen. Monostotic lesions may be more difficult to distinguish from other potential disorders, and additional imaging with computed tomography (CT) or magnetic resonance imaging (MRI) can be helpful, particularly in craniofacial cases. CT can provide additional detail illustrating the characteristic sclerotic and lytic changes and ground glass appearance seen within FD lesions (Fig. 18.3). MRI can add complementary information regarding content, size, and shape of lesions and can also be used to visualize important nearby neurovascular structures in craniofacial cases [22]. In addition, given the underlying pathophysiology of increased osteoblast activity, FD lesions are easily visualized on nuclear medicine bone scans, with areas of increased uptake corresponding to each bony lesion (Fig. 18.4). This type of imaging can be helpful to localize all potential affected areas and distinguish monostotic and polyostotic forms [23].
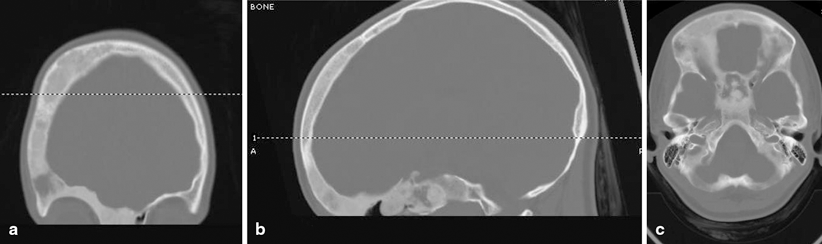
Fig. 18.3
Head CT images in a patient with craniofacial FD. Images reveal heterogeneous appearance of the skull with both sclerotic, as well as lytic changes and the characteristic a ground glass appearance. Multiple bones are involved, including right frontal bone, the body of sphenoid, the right pterygoid bone, the medial right orbital wall, the ethmoidal bone, the ethmoid air cells, the right orbital roof, and the medial aspect left frontal bone
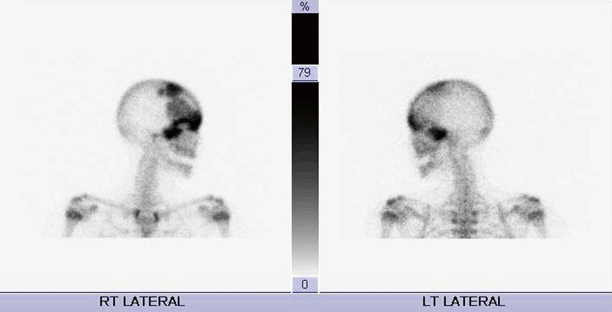
Fig. 18.4
Radionuclide bone scan in a patient with craniofacial FD. Images reveal increased uptake in multiple sites corresponding to FD lesions
Pathology
In most cases, a biopsy of the bony lesion is not required to make the diagnosis of FD, since characteristic findings on imaging can be diagnostic. Biopsy can potentially contribute to pathologic fracture and, therefore, should be performed with caution and only in situations where the diagnosis remains unclear, after appropriate imaging and consultation with expert radiologists have been pursued.
Pathologic findings pathognomonic for FD include the accumulation of fibrous tissue within the bone marrow, associated with abnormal osteoblasts shaped like immature spindle fibroblast-like cells arranged in parallel arrays or whirls (Fig. 18.5 and 18.6). Lesions typically expand from the medullary cavity outward to the cortical bone. Sequence analysis of the GNAS gene from a sample of the affected bone can confirm the disease-causing mutation [24].
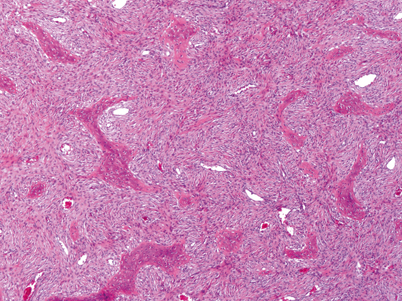
Fig. 18.5
Representative histological image. The tumor is composed of well-differentiated cords and sheets of spindle shaped fibroblast-like cells, which have been shown to have osteoblast proteins by immunocytochemistry. Embedded within this tissue are irregular, mineralized, woven bone trabeculae. There are no cuboidal osteoblasts rimming the bone trabeculae. There are a few large vascular channels. The tumor cells immunohistochemically express perostin [25]. (Picture and description provided by Sanford I. Roth MD)
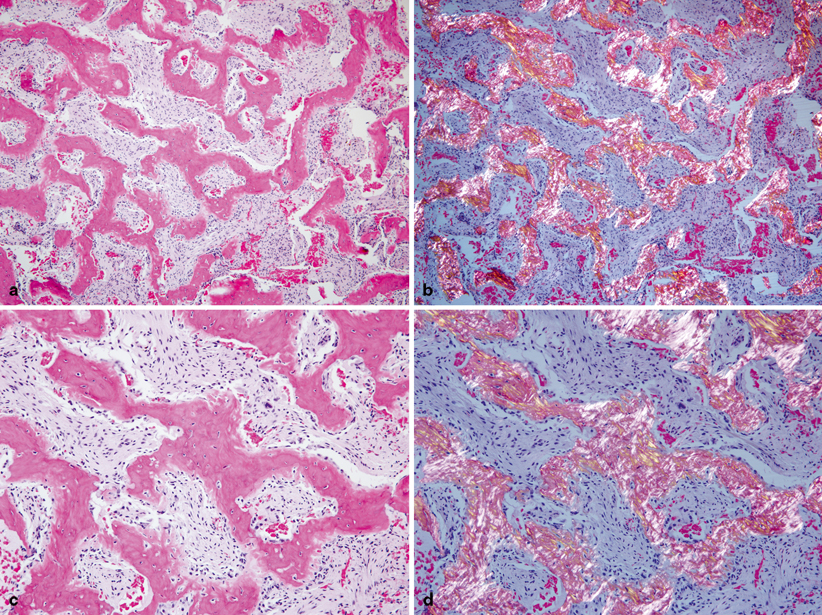
Fig. 18.6
Additional histological images. a Complex trabeculae of woven bone separated by a moderately cellular fibrous stroma. Anastomosing branching trabeculae of bone mimic Chinese characters. b Same image as a under polarizing lenses showing woven bone with absent lamellae. c At higher magnification, trabeculae of woven bone lack osteoblastic rimming. The fibrous stroma cells are bland, lacking nuclear hyperchromatism or pleomorphism. d Same image as c under polarizing lenses showing the characteristic woven bone with absence of osteoblastic rimming
Treatment
Medical Treatment
Medical therapy for all patients with FD includes optimizing factors known to affect their bone health. Most experts recommend that patients receive adequate amounts of daily vitamin D and calcium in order to prevent secondary hyperparathyroidism that can ultimately lead to concomitant rickets or osteomalacia and further weaken bones. In addition, subjects with renal phosphate wasting and hypophosphatemia may benefit from phosphate supplementation and calcitriol, although the efficacy of this approach has not been confirmed in clinical trials.
Nonpainful FD lesions that do not impinge on associated structures and are not associated with fracture or deformity can usually be followed expectantly. However, patients with FD can suffer significant pain related to the lesion itself or underlying pathologic fracture. Significant bone deformities caused by FD can also lead to disability and disfigurement. In addition, craniofacial lesions may expand to compromise the nearby important neurovascular structures. In these cases, additional medical therapy or surgical intervention may be indicated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


